
10 天前
10 天前
ICU里,每4个脓毒症患者就有1个会被急性肺损伤拖入鬼门关——肺部像被泼了强酸,肺泡壁破溃、水肿,氧气再也进不去血液。过去医生们总以为是免疫细胞失控引发的炎症风暴在捣鬼,直到武汉大学的研究团队揪出了藏在细胞核里的真凶:一个叫RUNX2的转录因子,它根本不需要免疫细胞帮忙,就能直接把肺上皮细胞推向死亡。更关键的是,这条通路的核心居然和我们细胞里的“铁仓库”有关。为什么脓毒症专挑肺下手?我们得从细胞里的“铁转运站”说起。
你可以把肺上皮细胞想象成肺泡的“城墙砖”,而线粒体就是砖里的“能量工厂”,工厂运转需要精准的铁供应——多了会炸,少了会停。
正常情况下,线粒体铁转运蛋白MFRN2是“送铁工”,它把适量的铁送进线粒体,然后会被泛素标记“回收”,维持铁的动态平衡。但当脓毒症的“信号弹”LPS(脂多糖)袭来时,细胞核里的RUNX2会突然活化,像拿到了总统令的官员,直接冲到USP16基因的启动子上,命令它大量生产USP16蛋白。
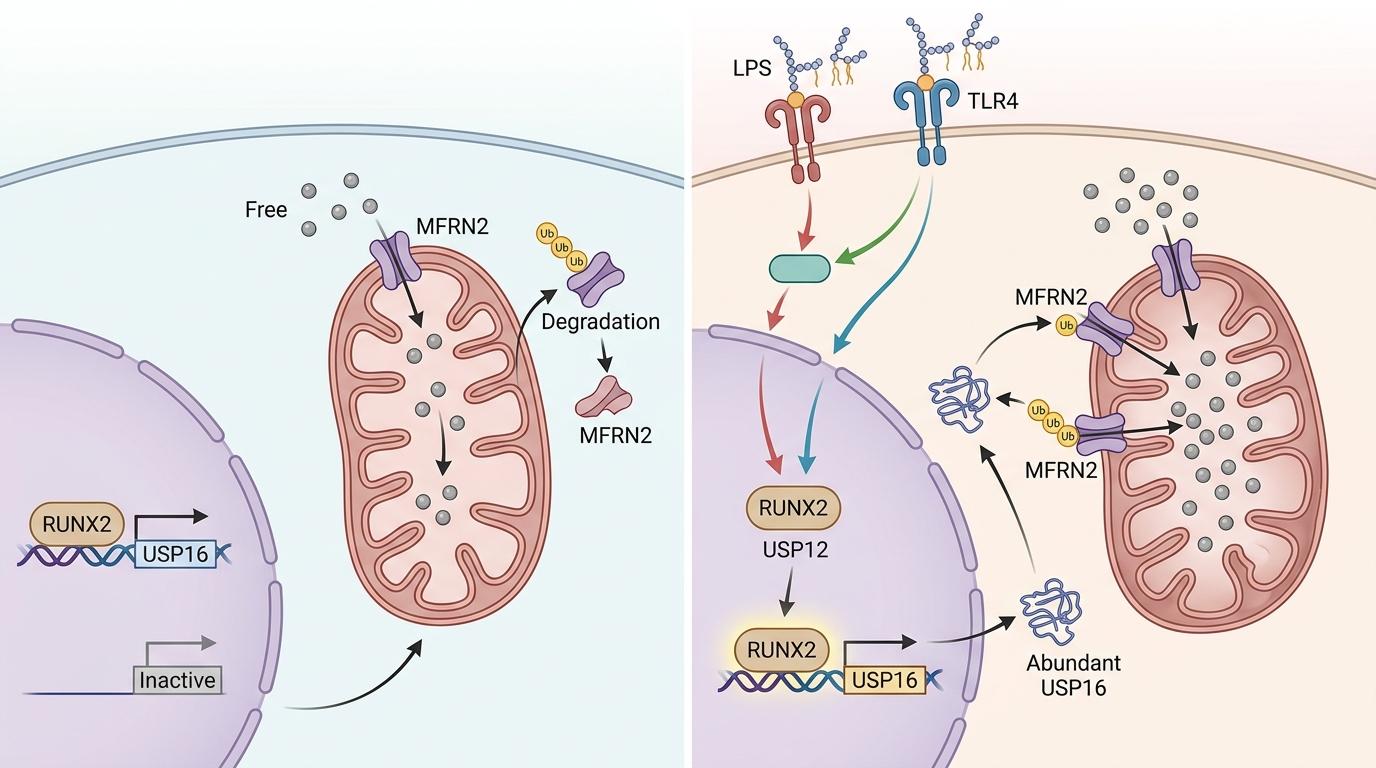
USP16是个“拆标记的工人”,它会把MFRN2身上的泛素标记拆掉,让这个“送铁工”一直留在岗位上。结果就是线粒体里的铁越堆越多,过量的铁催化出大量活性氧,像无数小炸弹炸碎细胞膜——这就是铁死亡,一种比凋亡更残酷的细胞死亡方式。
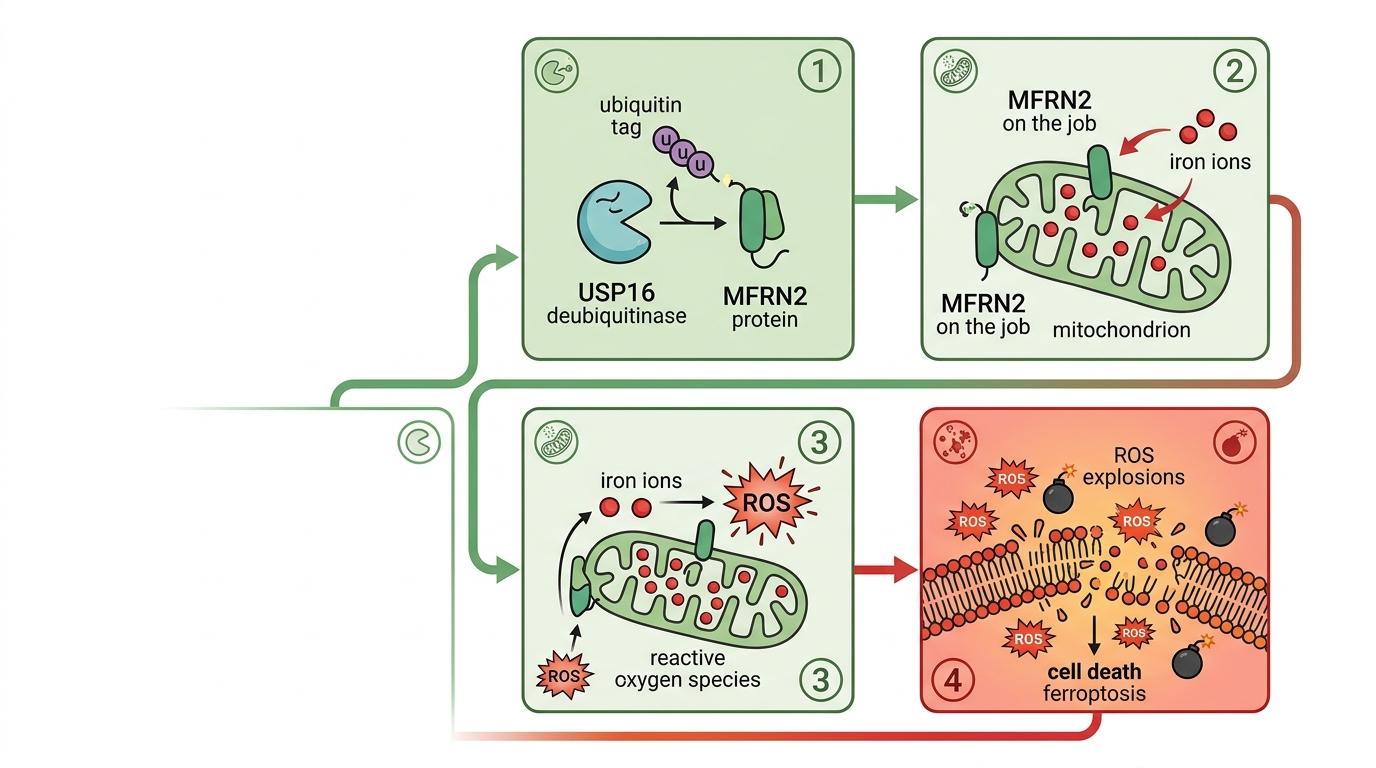
直给逻辑链: LPS刺激 → RUNX2活化 → 转录激活USP16 → USP16去泛素化稳定MFRN2 → 线粒体铁过载 → 铁死亡 → 肺上皮屏障破溃
就在RUNX2忙着下达死亡指令时,细胞里还有个天然的“刹车”——芳香烃受体AHR。它像个潜伏的督查,能直接和RUNX2结合,把它的“总统令”给扣下来,不让它去激活USP16。
研究团队在小鼠实验里验证了这个机制:当AHR过表达时,RUNX2的活性被抑制,MFRN2的泛素标记得以保留,线粒体铁稳态恢复,肺上皮细胞的铁死亡和凋亡都显著减少。更有意思的是,这个“刹车”还能和其他通路联动,比如抑制NF-κB的炎症信号,相当于同时给炎症和铁死亡踩了刹车。
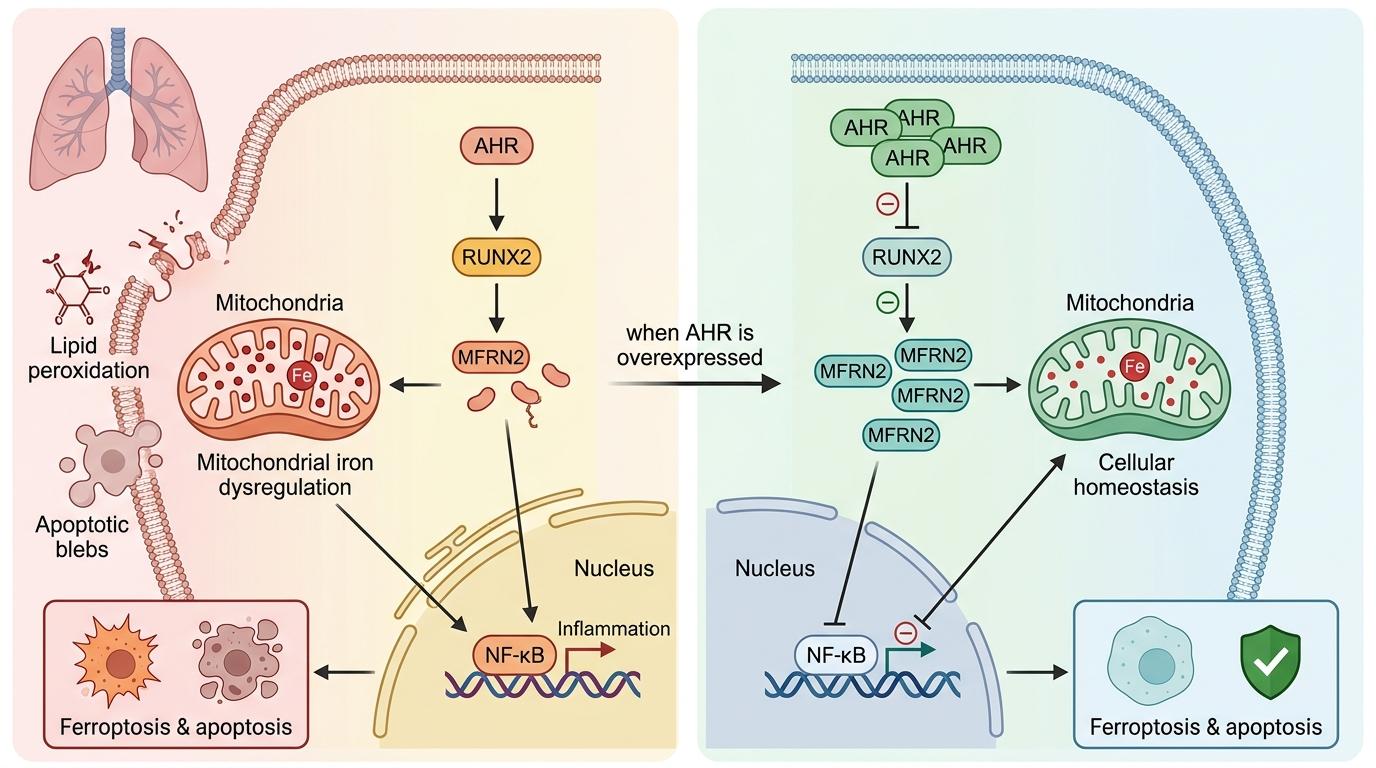
但这个“刹车”也有弱点:当LPS浓度过高时,AHR的抑制作用会被削弱,RUNX2就会重新掌权。这也解释了为什么脓毒症越严重,肺损伤越难逆转——我们体内的天然防御机制被冲垮了。
更值得关注的是,这个通路不仅存在于脓毒症肺损伤中,在肺癌、肺纤维化等疾病里,RUNX2的过度活化也扮演着类似的角色。它就像个细胞内的“反派开关”,一旦被打开,就会启动不同的损伤程序。
现在的问题是,我们能不能把这个发现变成治疗手段?
目前的思路有三个方向:一是直接抑制RUNX2的转录活性,比如用小干扰RNA或者小分子抑制剂,阻止它去激活USP16;二是增强AHR的“刹车”功能,比如用天然配体(如蔬菜里的吲哚类化合物)或者人工合成的AHR激动剂;三是直接靶向铁死亡,比如用铁螯合剂减少线粒体铁过载,或者用铁死亡抑制剂ferrostatin-1阻断脂质过氧化。
但这些思路都还在实验室阶段。比如RUNX2在骨发育中也有重要作用,直接抑制可能会导致骨质疏松等副作用;AHR的配体特异性很强,一不小心可能会激活其他通路。最现实的是,我们需要找到能特异性针对肺上皮细胞的递送系统,让药物只在肺部发挥作用,不影响其他器官。
不过,这个研究至少给了我们一个全新的方向:过去我们总在盯着免疫细胞的炎症风暴,现在发现肺上皮细胞自己也会“自杀”——治疗脓毒症肺损伤,不仅要“抗炎”,还要“保细胞”。
当我们把视线从宏观的炎症风暴拉回到微观的细胞通路,会发现人体的防御机制远比想象中精密:一个转录因子的活化,一个蛋白的泛素化修饰,就能决定一个细胞的生死,进而影响一个人的命运。
“铁稳则细胞安,细胞安则肺腑宁”,这或许是这个研究给我们最朴素的启示。未来的某天,当ICU里的脓毒症患者用上靶向RUNX2的药物时,我们会想起今天这个发现——原来藏在细胞核里的小小通路,能撬动生死的天平。而科学的意义,就是把这些隐秘的通路一个个找出来,变成照亮生命的光。
点击充电,成为大圆镜下一个视频选题!