对抗知识焦虑,从看懂这条开始
App 下载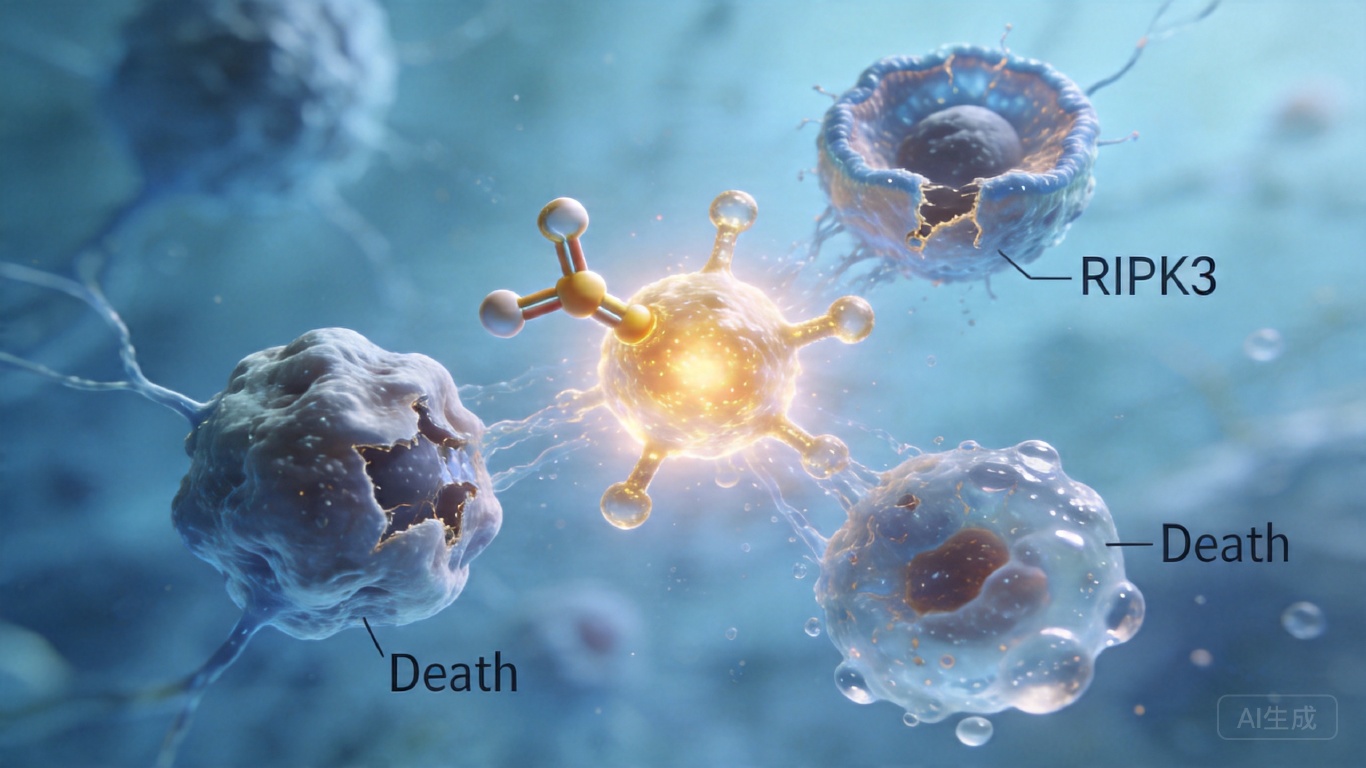
RIPK3:一个细胞死亡的全能调度员
MLKL蛋白|混合死亡模式|细胞死亡通路|RIPK3蛋白|分子细胞生物学|生命科学
对抗知识焦虑,从看懂这条开始
App 下载MLKL蛋白|混合死亡模式|细胞死亡通路|RIPK3蛋白|分子细胞生物学|生命科学
你或许从未想过,细胞的死亡也分“工种”——有的安静凋亡“体面退场”,有的爆裂焦亡“释放信号”,有的坏死凋亡“引发炎症”。过去科学家一直认为这些死亡路径各走各的,直到上海交通大学的团队发现了一个打破规则的“调度员”:RIPK3。它能在同一个细胞里同时启动三种死亡程序,把原本各不相干的死亡通路拧成一股绳,制造出一种全新的“混合死亡”模式。这不仅推翻了我们对细胞死亡的传统认知,更意味着我们找到了调控炎症和肿瘤的全新靶点。
RIPK3原本是坏死性凋亡这条“生产线”上的关键工人——它会给蛋白MLKL打上磷酸化标记,让MLKL聚集到细胞膜上钻洞,最终导致细胞爆裂死亡。但这次研究人员发现,当RIPK3被直接激活时,它会先招募MLKL,再陆续拉来RIPK1、FADD和Caspase-8这些“凋亡工人”,在自己身上组装出一个混合复合物。
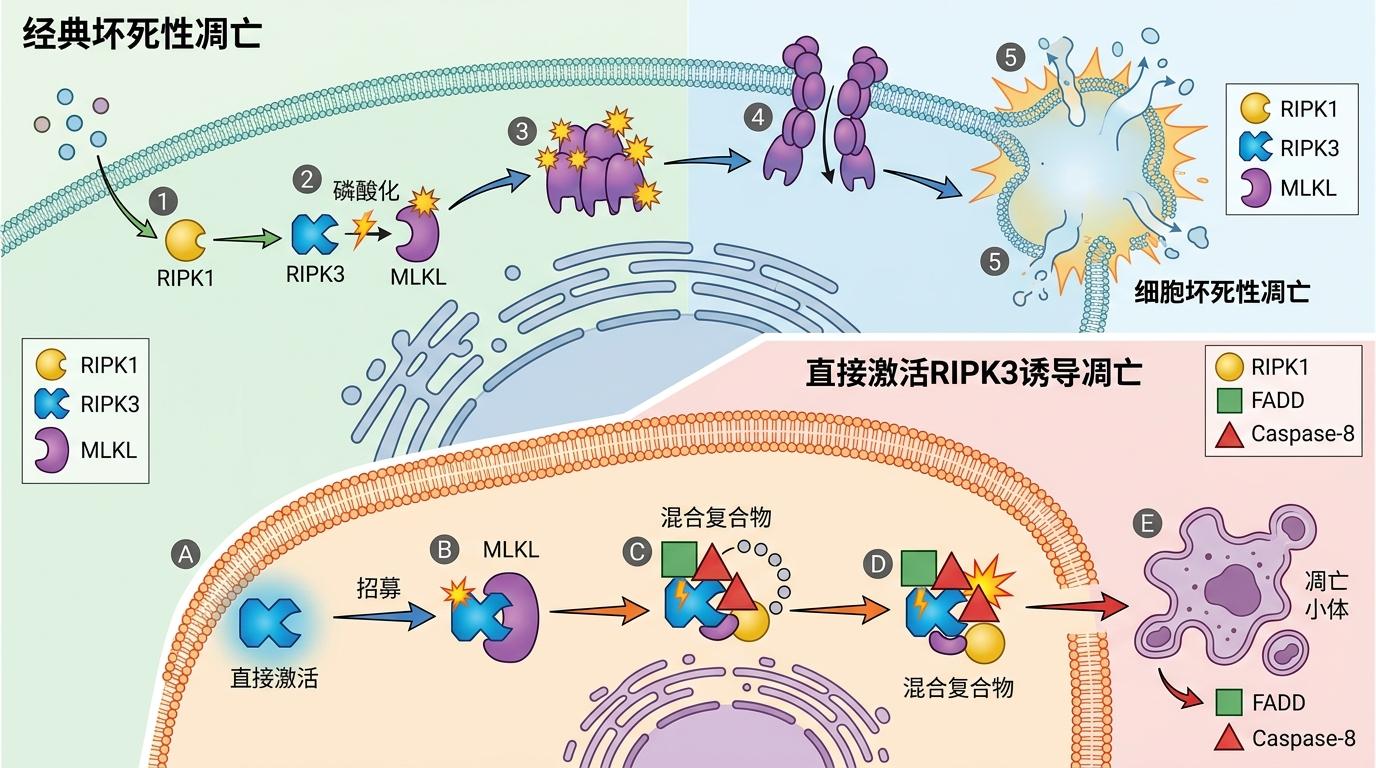
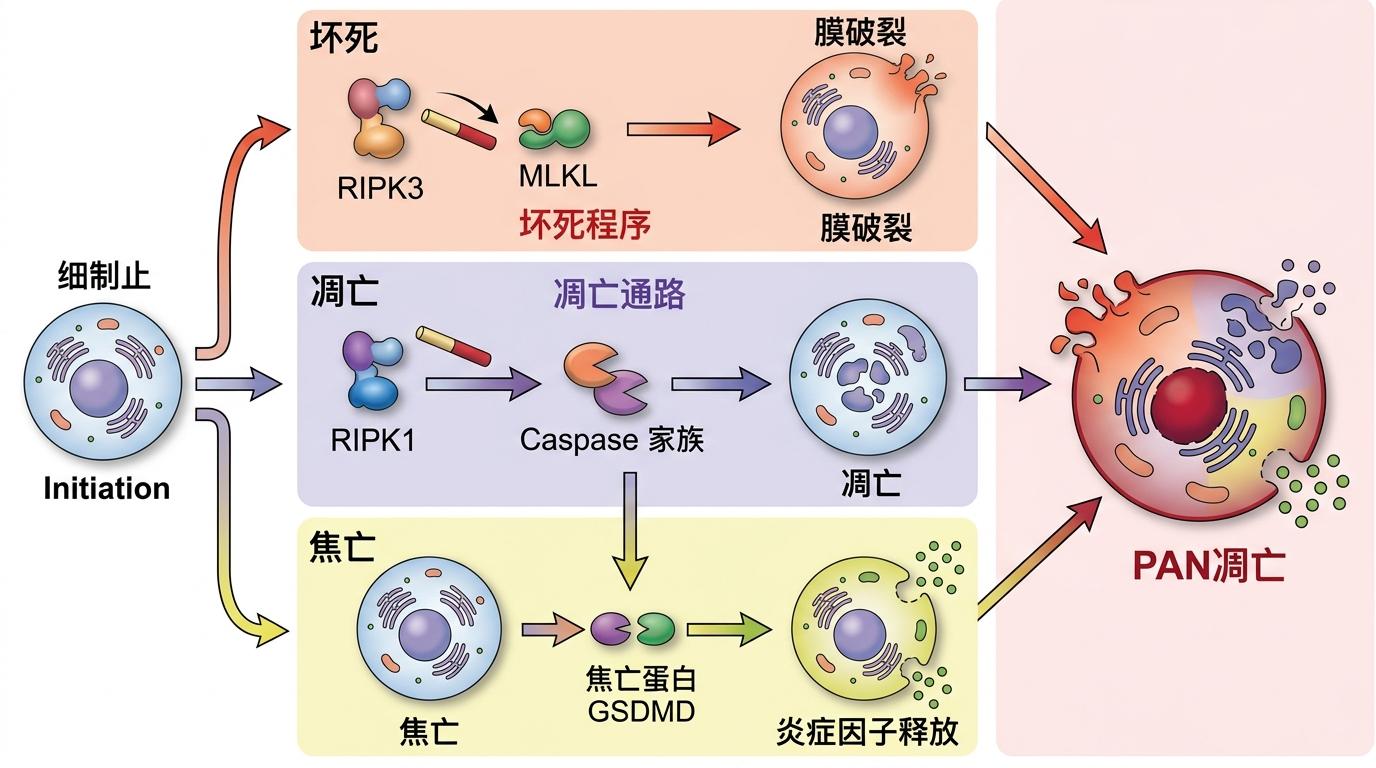
你可以把这个过程想象成一场接力赛:RIPK3先让MLKL跑第一棒启动坏死程序,接着RIPK1作为第二棒衔接凋亡通路,最后Caspase家族激活凋亡并切割焦亡蛋白GSDMD。三种死亡程序同时运转,让细胞既有凋亡的核碎裂,又有坏死的膜破裂,还能释放焦亡特有的炎症因子——这就是全新的PAN凋亡。
更精妙的是,这些“工人”对RIPK3的亲和力不同:MLKL喜欢和刚聚集成小团体的RIPK3结合,而RIPK1只对壮大后的RIPK3聚集体感兴趣。这种“按序入场”的机制,确保了三条通路不会互相干扰,又能协同起效。
PAN凋亡不是简单的“1+1+1”,三条通路之间还藏着复杂的制衡机制。比如Caspase-3会切割RIPK3和GSDMD,抑制坏死和焦亡的膜破裂;而RIPK3的激酶活性又会阻碍RIPK1的招募,减弱凋亡的强度。这种“此消彼长”的调控,让PAN凋亡释放的损伤信号和趋化因子,和单一死亡通路完全不同。
研究人员做了个对比:经典坏死性凋亡会大量释放ATP,而PAN凋亡释放的ATP量只有前者的三分之一;但它分泌的趋化因子CXCL10,却是经典凋亡的五倍之多。这些独特的信号分子,会吸引不同的免疫细胞——比如更多的T细胞被招募过来,这意味着PAN凋亡能更有效地激活抗肿瘤免疫。
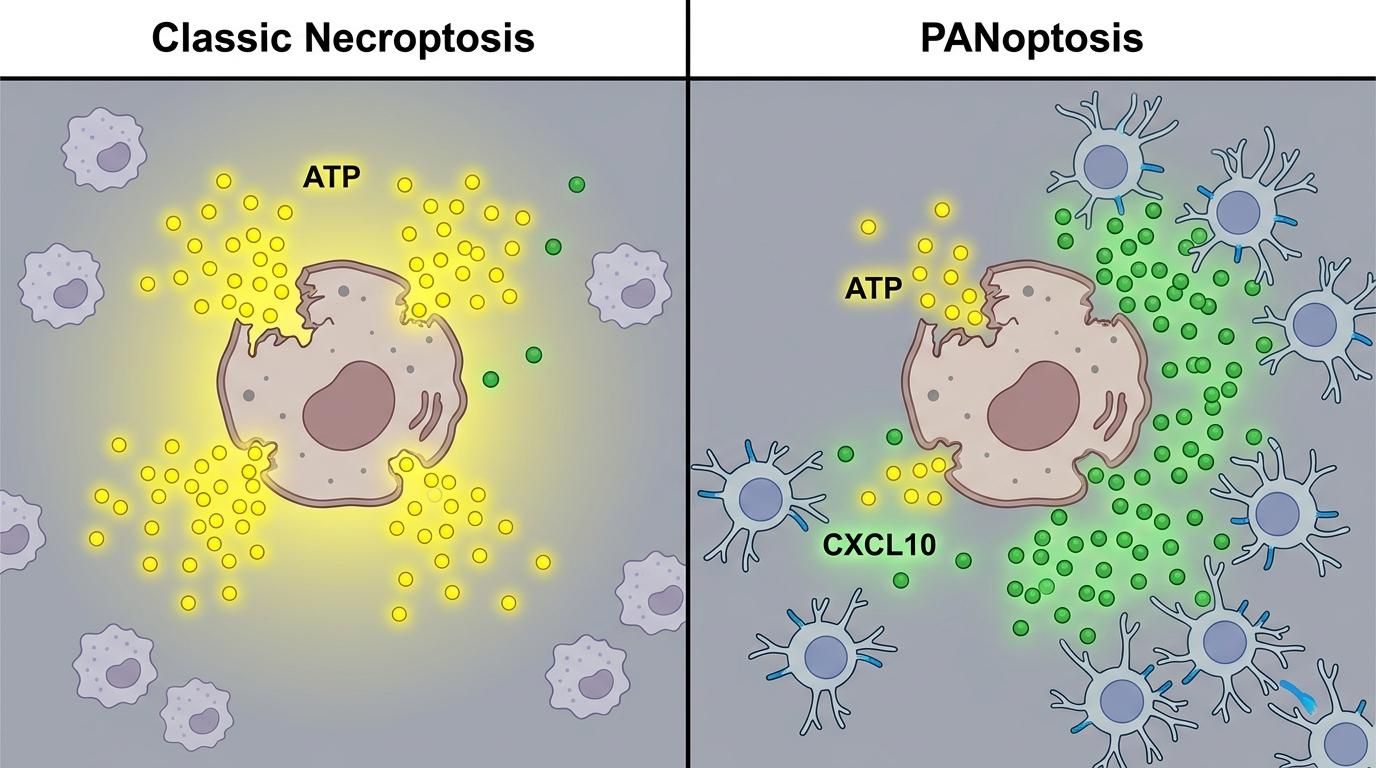
有意思的是,这种制衡还能防止死亡程序“过火”。如果RIPK3的激酶活性太强,就会抑制凋亡通路,导致细胞过度坏死引发炎症风暴;反过来,如果凋亡通路太活跃,又会切割RIPK3,阻断坏死和焦亡。这种动态平衡,让细胞能根据外界刺激调整死亡方式。
这个发现最吸引人的地方,在于它的临床潜力。在炎症性疾病中,比如急性肺损伤或炎症性肠病,过度的PAN凋亡会导致组织损伤,我们可以通过抑制RIPK3的聚集,来减少混合死亡的发生;而在肿瘤治疗中,很多癌细胞会对单一的凋亡或坏死治疗产生耐药性,我们可以激活RIPK3诱导PAN凋亡,让癌细胞无路可逃。
目前已经有研究显示,恢复肿瘤细胞中沉默的RIPK3表达,能让原本对免疫治疗不敏感的肿瘤,重新对PD-1抑制剂产生响应。这是因为PAN凋亡释放的独特信号,能打破肿瘤微环境的免疫抑制,让免疫细胞重新识别并攻击肿瘤。
不过现在还面临不少挑战:比如如何精准激活肿瘤细胞的RIPK3,同时不影响正常组织;不同细胞类型中RIPK3的调控机制是否存在差异。但可以确定的是,RIPK3这个“全能调度员”,已经为我们打开了一扇调控细胞命运的新大门。
过去我们总把细胞死亡看作单一的“终点”,但RIPK3的发现让我们意识到,死亡更像是一个复杂的“信号网络”。它不仅是细胞的终结,更是启动免疫反应、维持机体平衡的关键节点。
“死亡不是终点,而是新信号的起点。”这句话或许能概括这个发现的核心意义。未来当我们精准调控RIPK3这个枢纽时,或许就能让细胞死亡从“失控的破坏者”,变成“可控的治疗工具”——这不仅是细胞死亡研究的新纪元,更是精准医学的全新开端。