
9 天前
9 天前
五年生存率不到10%,手术加化疗也难撑过一年——这是被称为「癌王」的胰腺导管腺癌(PDAC)的残酷现实。更让人困惑的是,超过95%的它携带KRAS基因突变,而这种突变本该让癌细胞对铁死亡极度敏感。铁死亡,是一种铁依赖性的细胞死亡,核心是脂质过氧化物爆发式积累撑破细胞膜,恰恰是科学家为KRAS突变肿瘤量身定制的「死亡陷阱」。
但胰腺癌偏不按剧本走。它对铁死亡诱导剂的抵抗性,成了肿瘤生物学领域悬而未决的谜。直到最近,路德维希癌症研究所的团队在《Molecular Cell》上发表的研究,终于揪出了这场逃生大戏的总导演——缺氧诱导因子2(HIF-2)。
研究人员最初的假设很直接:既然肾癌细胞里高表达的HIF-2会让它们对铁死亡敏感,那同样高表达HIF-2的缺氧胰腺癌细胞,应该也逃不过铁死亡。
这个假设错了。
在缺氧条件下培养的胰腺癌细胞,反而对经典铁死亡诱导剂erastin和RSL-3更不敏感。更夸张的是,当他们用模拟胰腺肿瘤间质液的培养基,再叠加缺氧环境,癌细胞几乎对高剂量的erastin「无动于衷」。
原来,胰腺癌的两个标志性特征——间质液的独特代谢物组成和严重缺氧,在HIF-2的指挥下形成了协同防御。你可以把这个过程想象成:癌细胞在缺氧的「暗室」里,靠间质液提供的特殊「补给」,给自己焊上了一层不怕氧化的「防弹衣」。
铁死亡的核心是脂质过氧化物的积累,而细胞的保命武器是谷胱甘肽——一种强抗氧化剂——和它的搭档GPX4酶,能把过氧化的脂质还原回去。HIF-2做的,就是从「补弹药」和「掐火源」两个维度,强化这套保命系统。
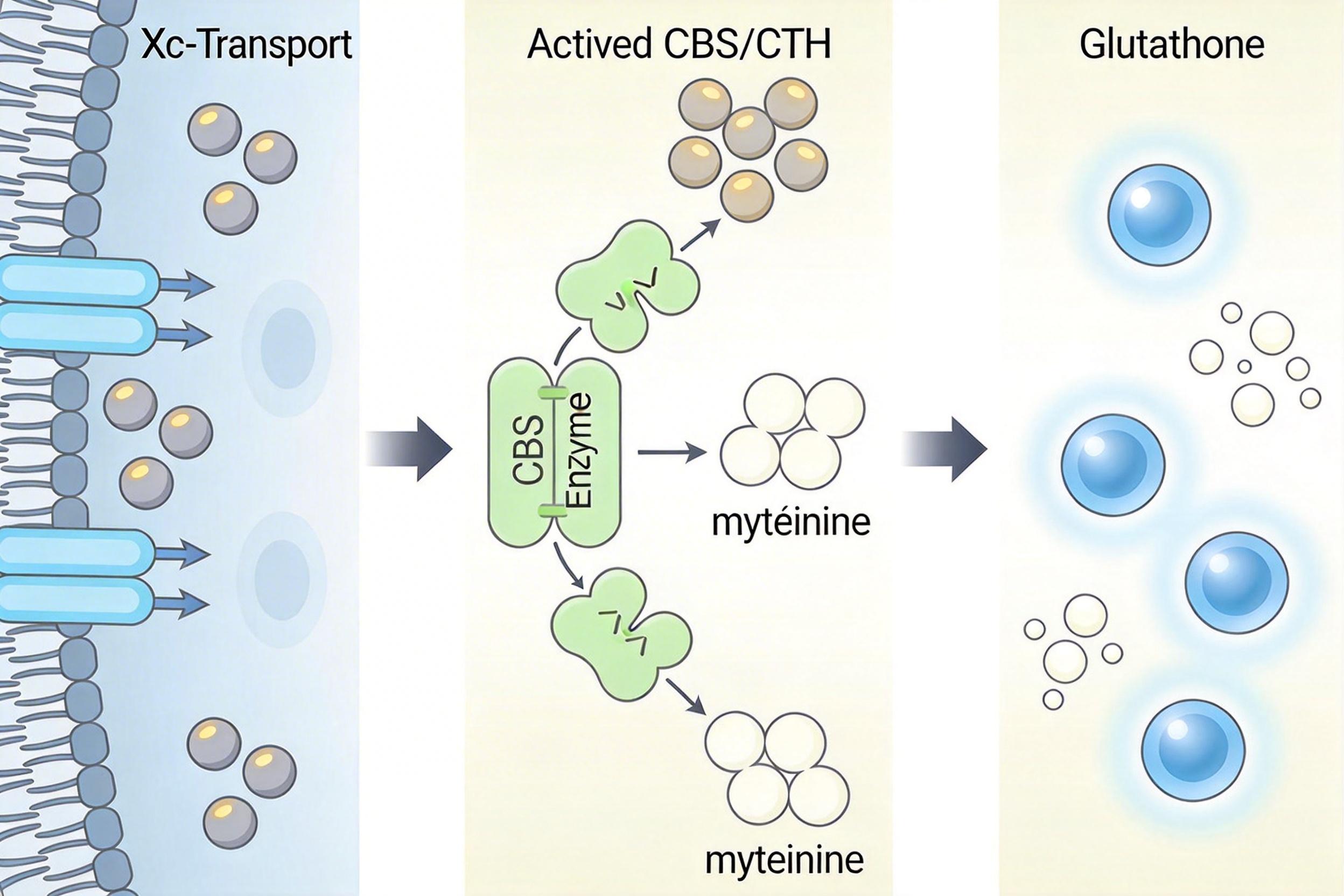
HIF-2的第一招,是给抗氧化系统「补够原料」。
它上调了系统Xc-胱氨酸转运体的表达,同时激活转硫途径的关键酶CBS和CTH——前者负责把胱氨酸「运进」细胞,后者能把细胞内的蛋氨酸转化为半胱氨酸。这双管齐下,让细胞内半胱氨酸的供应量大幅提升,而半胱氨酸正是合成谷胱甘肽的核心原料。相当于给细胞的「灭火队」源源不断送去灭火剂,让脂质过氧化物刚冒头就被消灭。
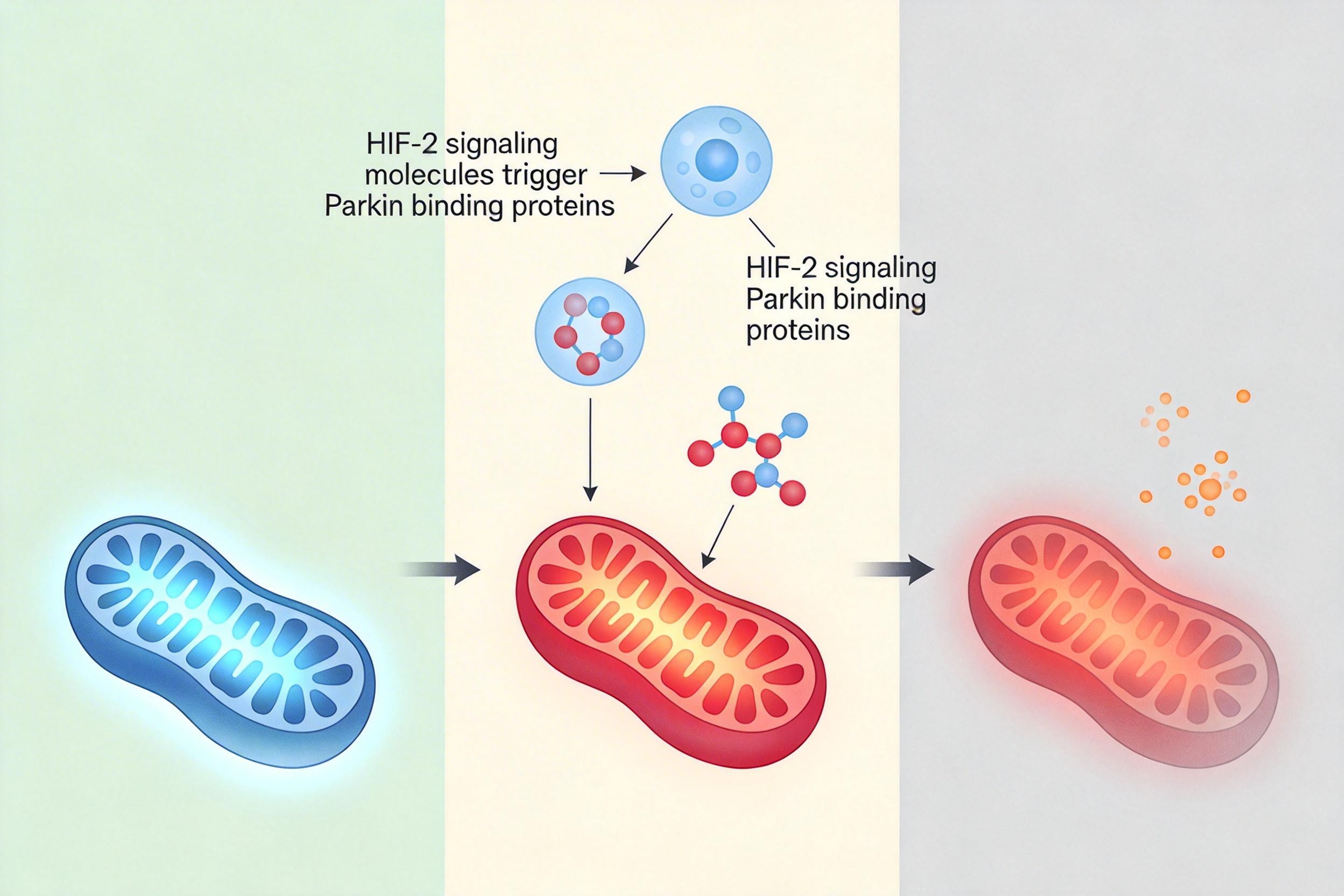
第二招,是直接掐灭产生过氧化物的「火源」。
HIF-2会诱导Parkin介导的线粒体自噬——有选择地清除那些功能异常的线粒体。线粒体是细胞内活性氧(ROS)的主要来源,而ROS正是催生脂质过氧化物的「火种」。把有问题的线粒体清掉,相当于拔掉了火灾的导火索,从根源上减少了过氧化物的产生。
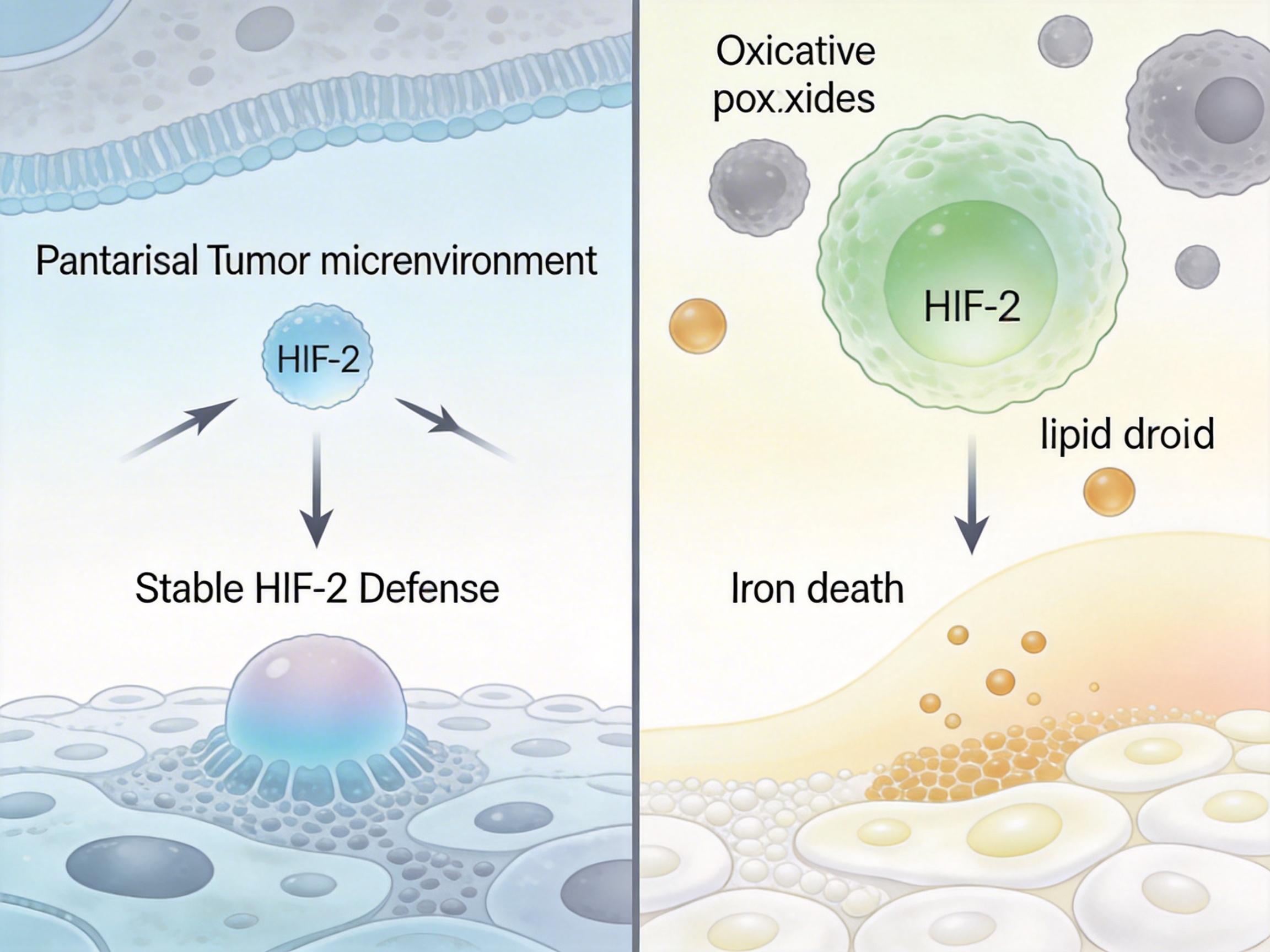
更关键的是,这套防御机制只在胰腺癌的特殊微环境里生效。在肾癌细胞里,HIF-2会调控脂质代谢,让细胞更容易积累过氧化物,反而成了铁死亡的「帮凶」。同一种分子,在不同肿瘤里扮演完全相反的角色,这正是肿瘤微环境的魔力——它能彻底改写基因的功能。
现在,科学家终于搞懂了胰腺癌抵抗铁死亡的核心机制,但要把这个发现变成患者能用的疗法,还有不少坎要过。
第一关是药物的特异性。HIF-2的抑制剂已经在肾癌中获批,但胰腺癌里的HIF-2不仅在癌细胞里表达,还存在于癌相关成纤维细胞中,后者会调控免疫抑制微环境。直接抑制HIF-2会不会影响正常细胞的氧感知功能?会不会破坏红细胞生成?这些都需要更细致的研究。
第二关是铁死亡诱导剂的体内难题。目前的铁死亡诱导剂要么难以穿透胰腺癌致密的间质,要么在体内容易被代谢分解,无法达到有效浓度。如何把药物精准递送到肿瘤内部,是绕不开的问题。
第三关是联合治疗的节奏。单独抑制HIF-2,或者单独诱导铁死亡,可能都不足以攻破胰腺癌的防线。但什么时候联合?用什么剂量组合?这些都需要在动物模型和临床试验里反复摸索。
不过,至少现在我们知道了方向——不是跟胰腺癌硬碰硬,而是先拆掉它的「防弹衣」。
过去我们总以为,癌细胞的耐药性是单个基因突变的结果,但胰腺癌的故事告诉我们,肿瘤从来不是孤立的细胞团,而是一个和微环境共生的「生态系统」。HIF-2就是这个生态系统里的「总调度」,它能根据环境信号,重新编排癌细胞的生存策略。
微环境塑造癌细胞,癌细胞反过来改造微环境——这才是「癌王」最可怕的地方。而破解这个谜题的意义,不仅在于找到胰腺癌的新靶点,更在于让我们意识到:对抗癌症,不能只盯着癌细胞本身,还要看清它赖以生存的「土壤」。
癌细胞的命运,从来不是基因写死的剧本。
点击充电,成为大圆镜下一个视频选题!