对抗知识焦虑,从看懂这条开始
App 下载
抑制锌转运,破解糖尿病细胞移植缺氧死局
同济大学|缺氧悖论|锌离子调控|β细胞凋亡|干细胞胰岛类器官|代谢内分泌疾病|医学健康
对抗知识焦虑,从看懂这条开始
App 下载同济大学|缺氧悖论|锌离子调控|β细胞凋亡|干细胞胰岛类器官|代谢内分泌疾病|医学健康
当医生把实验室培养的干细胞胰岛类器官植入糖尿病患者体内,最凶险的时刻不是手术台上,而是术后的前72小时。没有血管供血的类器官泡在缺氧的组织液里,90%的功能性β细胞会在一周内凋亡——这是困扰干细胞糖尿病治疗10年的「缺氧悖论」:缺氧本该触发血管生成信号自救,但干细胞胰岛的β细胞却会先被缺氧杀死,连求救信号都发不出来。2026年4月,同济大学生命科学团队在《Cell Stem Cell》发表的研究,找到了撬开这个死局的微小支点:锌离子。
你可以把干细胞胰岛的β细胞想象成一个精密的胰岛素加工厂,锌离子原本是车间里的「打包工人」——它和胰岛素结合形成六聚体,帮助胰岛素稳定储存。但同济团队的实验发现,干细胞分化来的β细胞里,锌离子会越攒越多,最终变成「车间里的废品堆」:过量锌会触发活性氧爆发,直接氧化损伤细胞内的能量传感器AMPK。
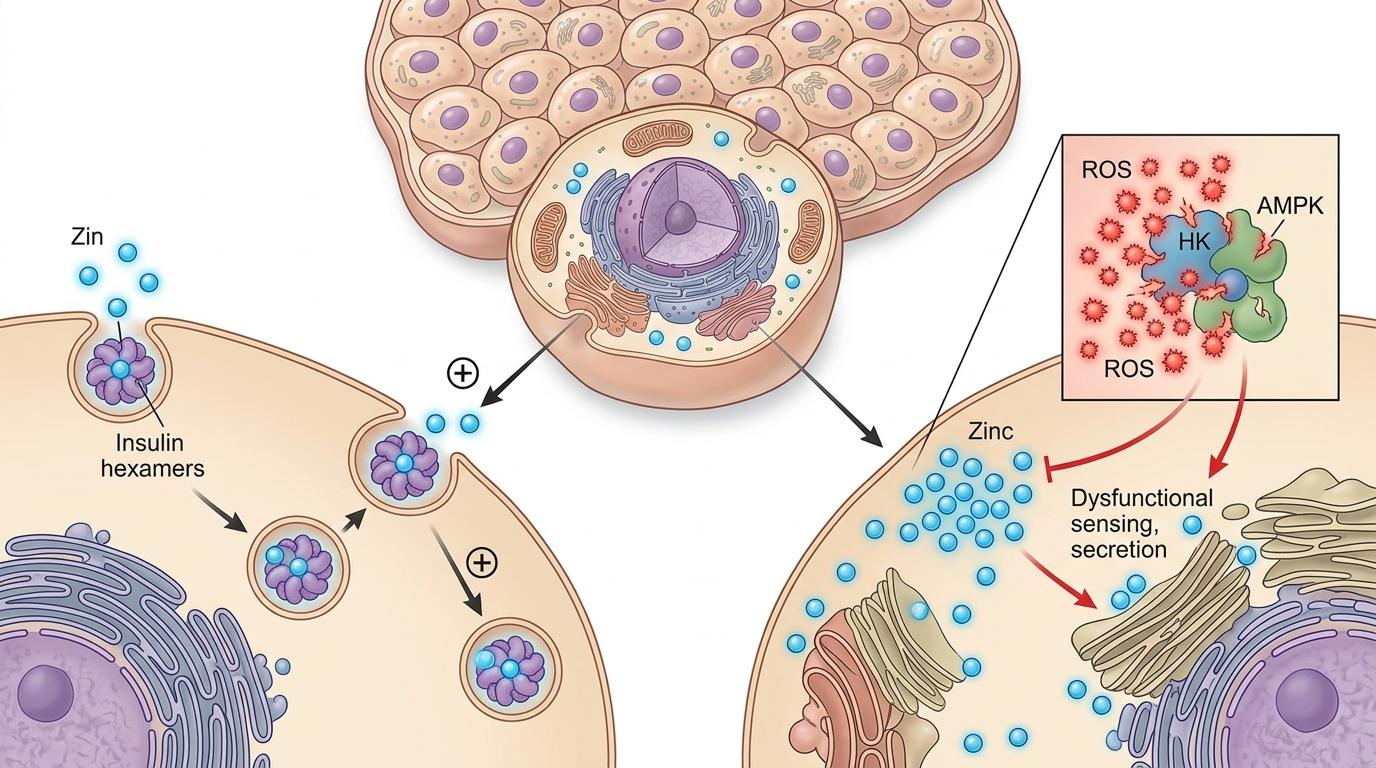
AMPK就像细胞的「应急总指挥」,一旦被氧化失活,β细胞不仅失去了应对缺氧的应激能力,连基本的胰岛素加工效率都会暴跌。实验数据显示,当用锌载体模拟锌过载时,干细胞胰岛的活性氧水平飙升3倍,AMPK的活性下降70%,成熟胰岛素颗粒的密度减少近一半。
更关键的是,这种锌过载导致的代谢脆弱性,让干细胞胰岛失去了缺氧时的自救本能:正常细胞会激活HIF1A因子启动血管生成信号,但锌过载的β细胞连启动信号的力气都没有,只能在缺氧环境里慢慢凋亡。
研究团队筛选了1685种小分子化合物,最终锁定了SU6656——一种能抑制锌转运蛋白ZnT8的化学物质。它的作用简单直接:把β细胞里堆成山的锌离子「运出去」,让AMPK重新苏醒。
苏醒的AMPK立刻启动了两条关键通路:一方面,它促进线粒体修复,提升β细胞的能量代谢效率,让干细胞胰岛的胰岛素分泌能力提升了2.3倍,甚至能和人类原代胰岛媲美;另一方面,它绕开了失效的HIF1A通路,直接激活ETS1转录因子,强制启动血管生成信号VEGFA。
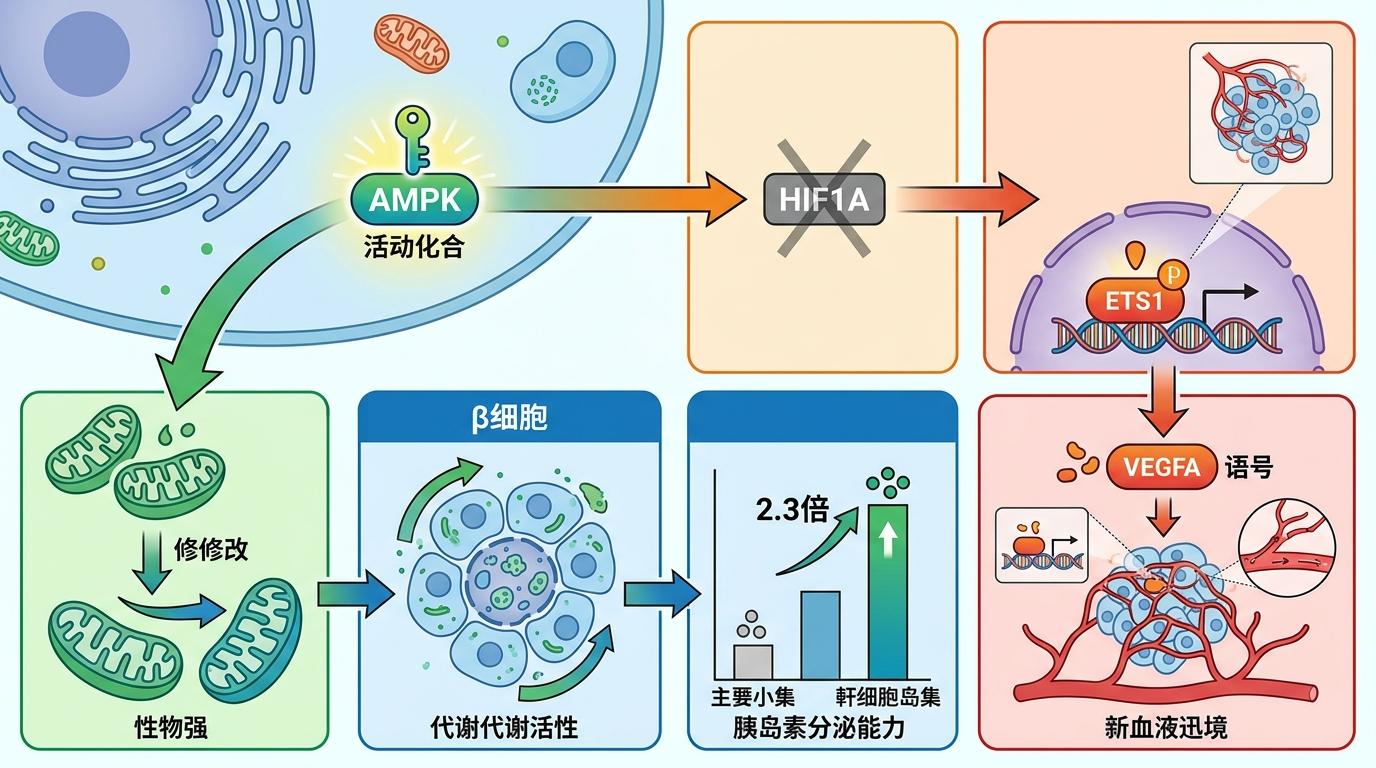
在1%氧浓度的体外缺氧实验中,SU6656处理过的干细胞胰岛,β细胞死亡率从45%降到了12%;移植到糖尿病小鼠体内后,移植物的血管化速度提升了3倍,小鼠的血糖在1周内就恢复了正常,且能稳定维持14周以上。
更值得关注的是,这个策略并非「拆东墙补西墙」:抑制锌转运不仅没有影响胰岛素的正常储存,反而因为β细胞代谢状态改善,胰岛素的成熟效率还提升了1.7倍。
当然,这个研究也并非完美的终极解决方案。目前的实验仅在小鼠模型中验证,要走向临床还需要解决三个核心问题:首先,SU6656的长期安全性需要验证,毕竟它原本是Src激酶抑制剂,长期使用可能存在脱靶风险;其次,干细胞胰岛的免疫排斥问题仍未解决,这个策略只提升了细胞的存活能力,没涉及免疫逃逸;最后,大规模生产时的锌转运调控精度需要控制,过少的锌会影响胰岛素储存,过多又会回到原来的死局。
但不可否认的是,它补上了干细胞糖尿病治疗中最关键的一块拼图:过去10年,科学家们要么在想办法让干细胞胰岛更成熟,要么在想办法让移植部位更快血管化,却没人注意到「锌过载」这个微小的环节。这个研究让我们意识到,有时候破解复杂的医学难题,不需要颠覆式的技术,只需要找到那个被忽略的「微小支点」。
当我们谈论糖尿病的治愈希望时,总习惯把目光投向那些宏大的技术——基因编辑、干细胞分化、免疫调控,但同济团队的研究却提醒我们:细胞的代谢平衡,往往藏在那些被忽略的微小分子里。
「精准调控代谢,比强行干预更有效。」这不仅是干细胞胰岛治疗的启示,也是整个再生医学的底层逻辑:我们不需要扮演上帝去创造完美的细胞,只需要学会倾听细胞的信号,帮它回到本该有的平衡状态。或许在不远的未来,糖尿病患者接受的不是一次复杂的移植手术,而是提前用锌调控预处理的干细胞胰岛,像植入一个能自动适应环境的「微型胰腺」,悄无声息地恢复血糖平衡。