
21 天前
21 天前
2026年3月,上海的实验室里,一群小鼠经历了一场生死考验:被注射致死剂量的内毒素后,普通小鼠在炎症风暴中陆续死亡,而携带特殊基因改造的小鼠,却有近80%活了下来。这不是科幻情节——这些小鼠的CYLD蛋白被修改了一个关键位点,能抵抗Caspase-8的切割。正是这个微小的分子变化,让它们在致命的脓毒症休克中活了下来。这背后,是科学家追踪了十几年的炎症开关:Caspase-8和CYLD的分子对决,终于露出了全貌。
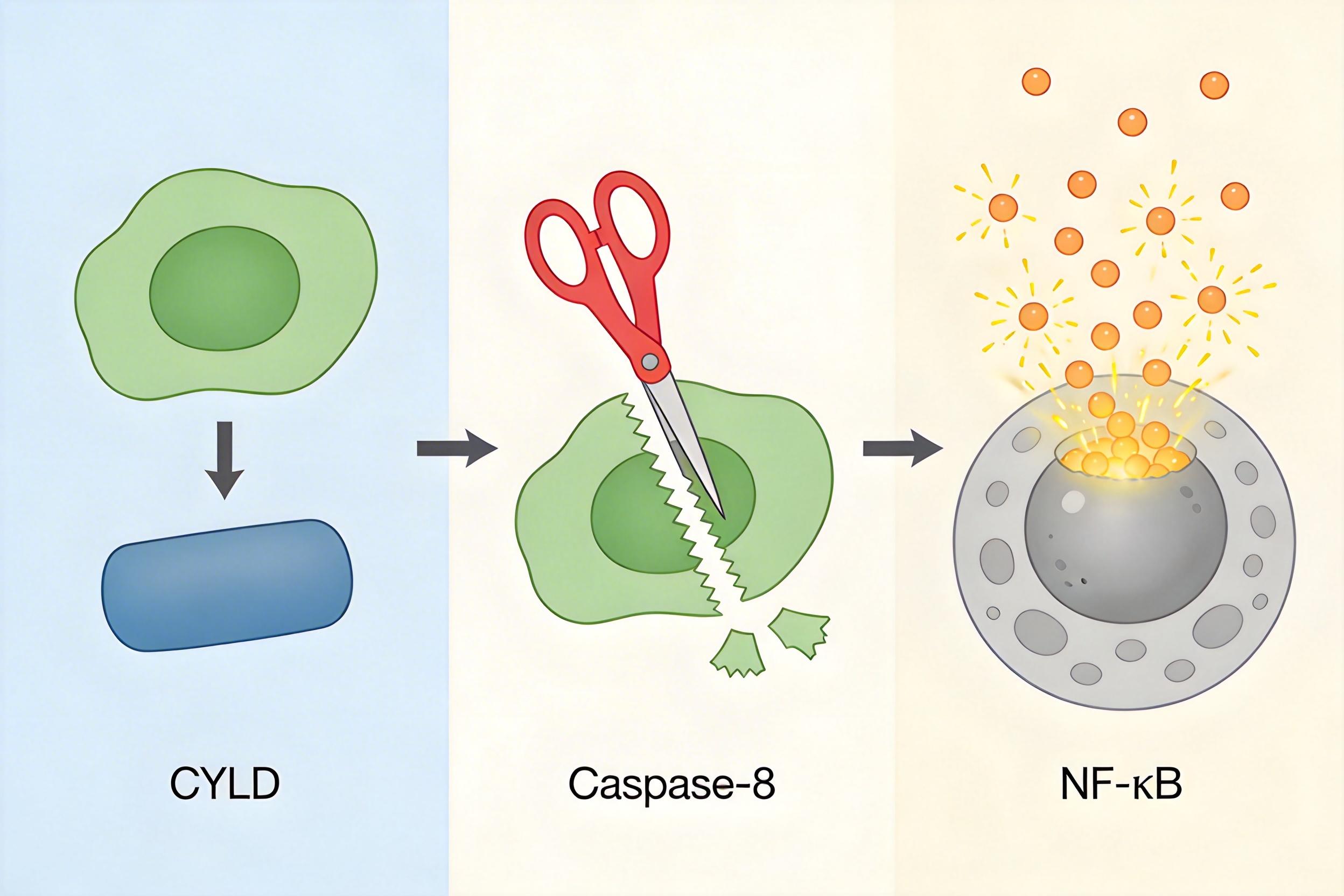
你可以把细胞里的炎症调控想象成一个带刹车的油门:NF-κB是油门,负责驱动炎症因子释放;CYLD就是刹车,通过去泛素化作用,把NF-κB的活性按下去。
但当细菌脂多糖(LPS)入侵时,Caspase-8会被激活——它像一把精准的剪刀,专挑CYLD蛋白上的D215位点切割。被剪碎的CYLD失去了去泛素化活性,刹车彻底失灵:NF-κB不受控制地涌入细胞核,指挥细胞疯狂生产炎症因子,最终引发全身性的炎症风暴,也就是脓毒症休克。
研究团队做了一组关键对照:在天生缺失Caspase-8的抗休克小鼠体内,再敲除CYLD基因,原本对LPS完全免疫的小鼠,立刻恢复了对脓毒症的敏感性。这直接坐实了CYLD的核心地位——Caspase-8正是通过摧毁CYLD,才打开了炎症的闸门。
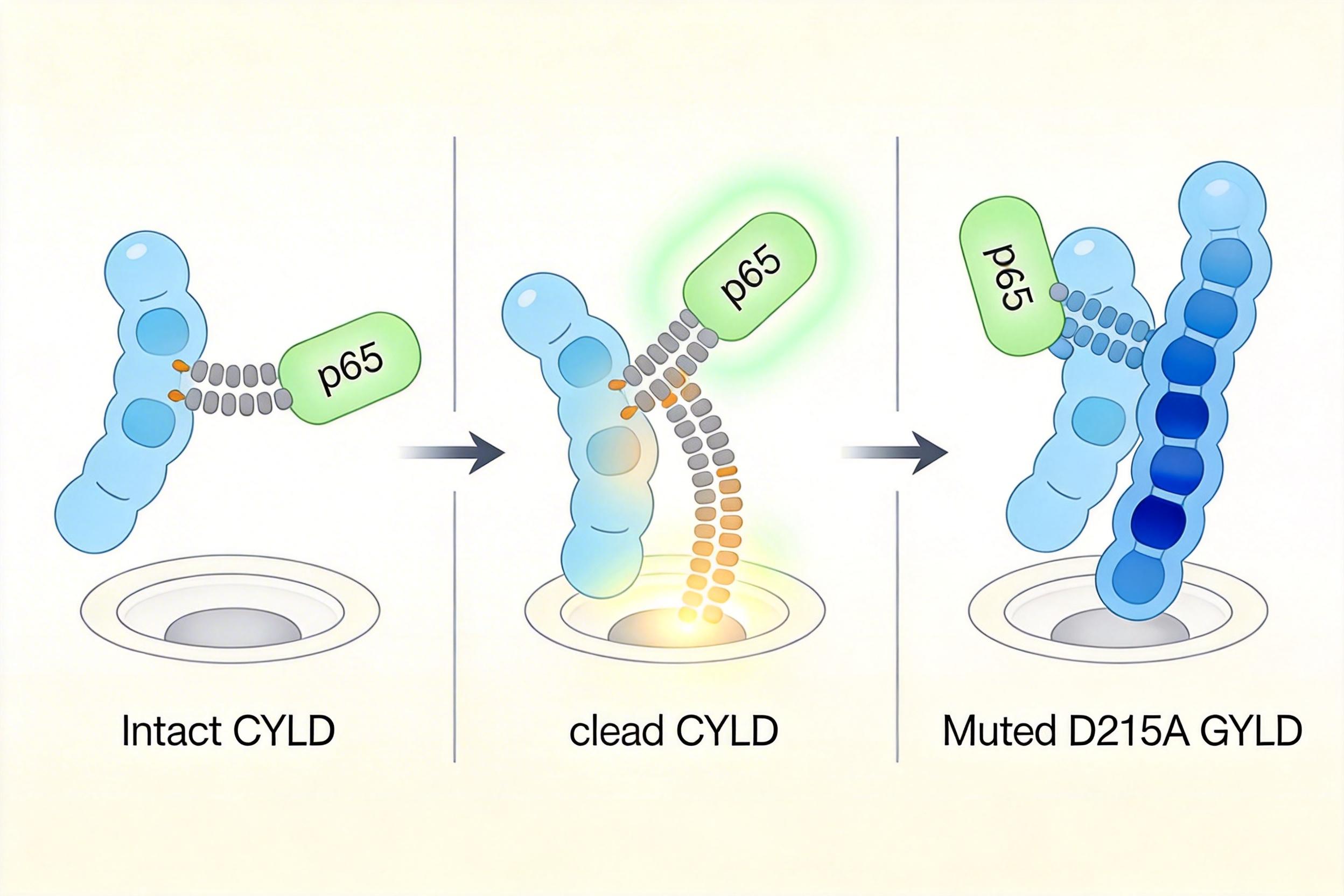
为了验证CYLD切割的关键作用,团队用基因编辑技术构建了CYLD D215A突变小鼠——把CYLD上被Caspase-8识别的切割位点,换成了一个“剪不动”的氨基酸。
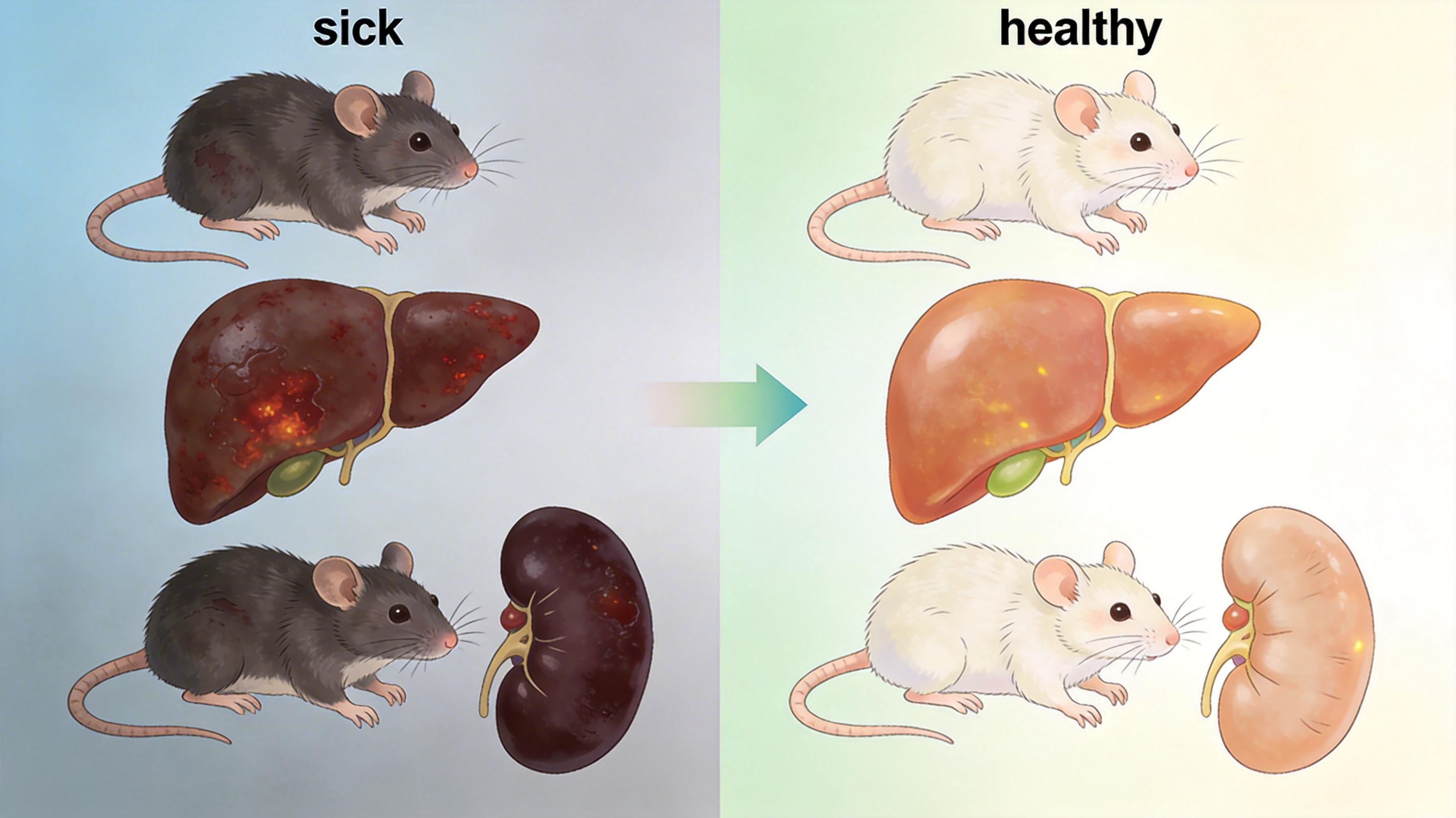
实验结果极具说服力:注射致死剂量LPS后,野生型小鼠在48小时内死亡率超过90%,而突变小鼠的死亡率不到20%。解剖显示,突变小鼠的肝脏、肾脏等器官几乎没有炎症坏死,血清里的TNF-α、IL-6等炎症因子水平,只有野生型的1/3。
更关键的机制也被揭开:CYLD原本能移除NF-κB亚基p65上的M1型线性泛素链,阻止它进入细胞核。当Caspase-8切割CYLD后,p65上的泛素链无法被移除,核转位效率提升了3倍,炎症信号被持续放大。而D215A突变让CYLD保持完整,牢牢按住了p65的“核转位开关”。
这项研究带来的不只是机制突破,还有两个明确的临床方向:
一是治疗靶点。既然阻断Caspase-8对CYLD的切割就能抗休克,那么开发能特异性结合CYLD D215位点的小分子抑制剂,或者改造CYLD使其抵抗切割,就能成为脓毒症的精准疗法。目前团队已经筛选出3个能稳定CYLD的先导化合物,在细胞实验中能将LPS诱导的炎症因子水平降低70%以上。
二是生物标志物。研究发现,Caspase-8切割CYLD产生的N端片段CP25,会通过TRIF/Caspase-8依赖的途径分泌到血清中。在脓毒症患者的血清样本中,CP25的浓度是健康人的6-10倍,且与疾病严重程度正相关。这意味着CP25可以作为脓毒症早期诊断的生物标志物,比目前常用的降钙素原提前12小时预警。
当然,挑战依然存在:Caspase-8同时还调控细胞凋亡,完全抑制它可能导致免疫缺陷。如何精准阻断它对CYLD的切割,而不影响其正常功能,是接下来的核心难题。
每年全球有超过1100万人死于脓毒症,其中很多人并非死于感染本身,而是死于失控的炎症风暴。过去几十年,科学家尝试过阻断单个炎症因子、抑制NF-κB活性,但都因副作用或效果不佳失败。
这次的研究,终于找到了炎症风暴的关键“开关”——不是某个孤立的因子,而是Caspase-8和CYLD之间的动态平衡。“守住CYLD,就守住了炎症的刹车”,这不仅是实验室里的分子发现,更是给数百万脓毒症患者带来的新希望。未来的抗炎治疗,或许不再是粗暴地抑制炎症,而是精准地调控平衡。
点击充电,成为大圆镜下一个视频选题!