对抗知识焦虑,从看懂这条开始
App 下载
从200个样本里,挖出蛋白质进化的最优解
高通量筛选|蛋白质序列优化|MULTI-evolve框架|Arc研究所|合成生物学|生命科学
对抗知识焦虑,从看懂这条开始
App 下载高通量筛选|蛋白质序列优化|MULTI-evolve框架|Arc研究所|合成生物学|生命科学
想象一下:一个由100个氨基酸组成的蛋白质,可能的序列组合多达20^100种——这个数字比可观测宇宙里的原子总数还要多。过去,科学家要从这天文数字的可能性里找功能更强的蛋白质,要么靠碰运气试错,要么得花数月做5到10轮实验筛选。但现在,Arc研究所的团队用一套叫MULTI-evolve的框架,只测200个样本,就能在数周里让蛋白质功能提升几十甚至上百倍。他们是怎么把大海捞针变成精准捕鱼的?
过去的机器学习模型,要么靠单突变数据训练,要么测几千个随机变体——前者不知道突变之间会怎么互相影响,后者大部分数据都是在教模型“什么是没用的”。就像你想学做一道好吃的菜,却对着一万份黑暗料理研究,最后只学会了避开错误,根本不知道真正的美味配方是什么。
MULTI-evolve的思路反了:先找到15到20个能增强蛋白质功能的单突变,然后系统测试这些突变的所有成对组合。这就像先挑出所有能提鲜的调料,再试哪两种搭配最惊艳——每一次测试都在教模型“什么是有效的协同”,而不是浪费精力在无效组合上。
他们用12个不同蛋白质家族的数据验证:哪怕把训练数据砍到10%,模型依然能准确预测5到7个突变组合的功能。核心就在于,这些成对组合的数据,能让模型抓住蛋白质里的“上位效应”——也就是突变之间1+1>2或者1+1<2的协同或拮抗作用,这才是多突变体功能提升的关键。
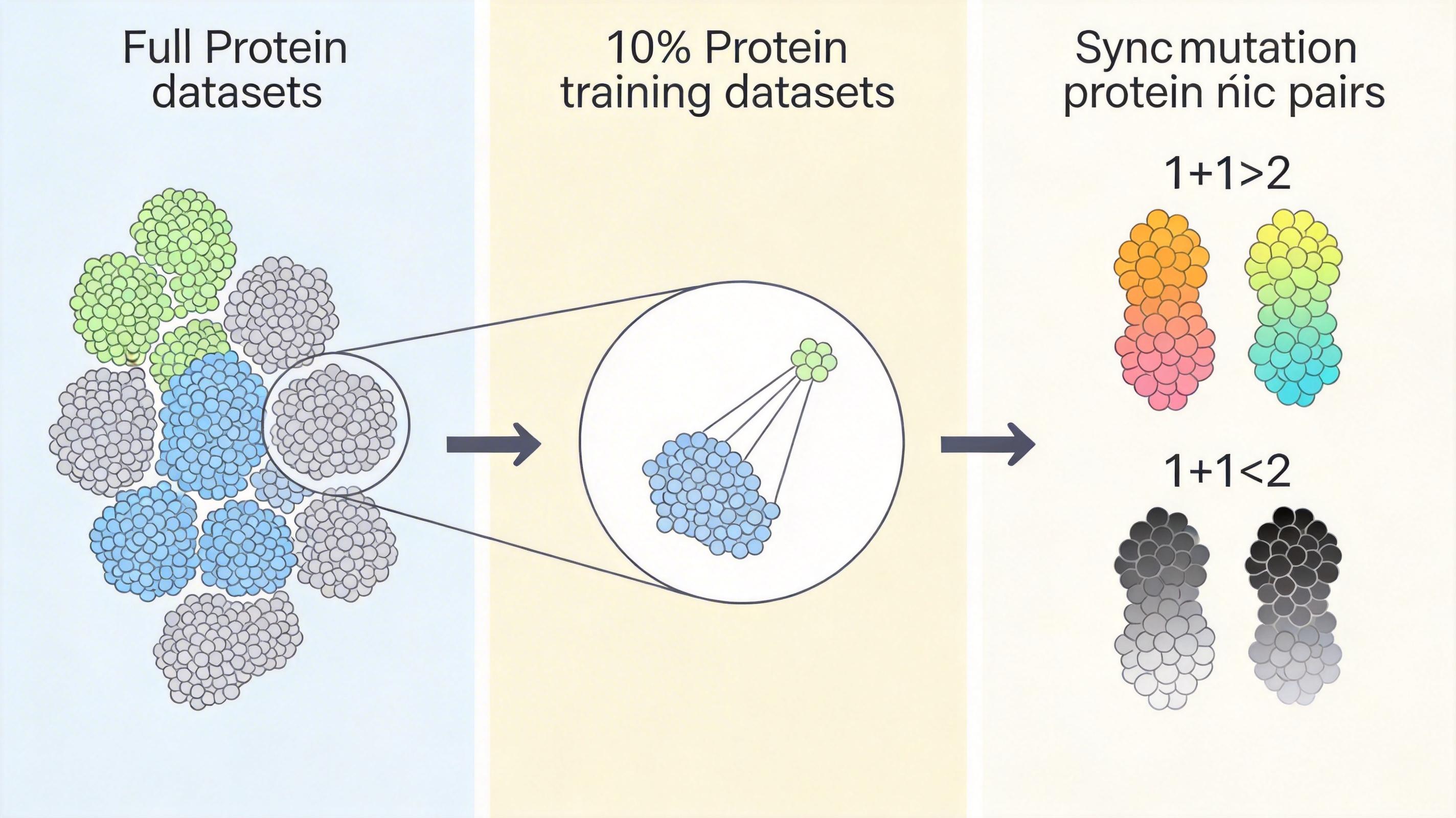
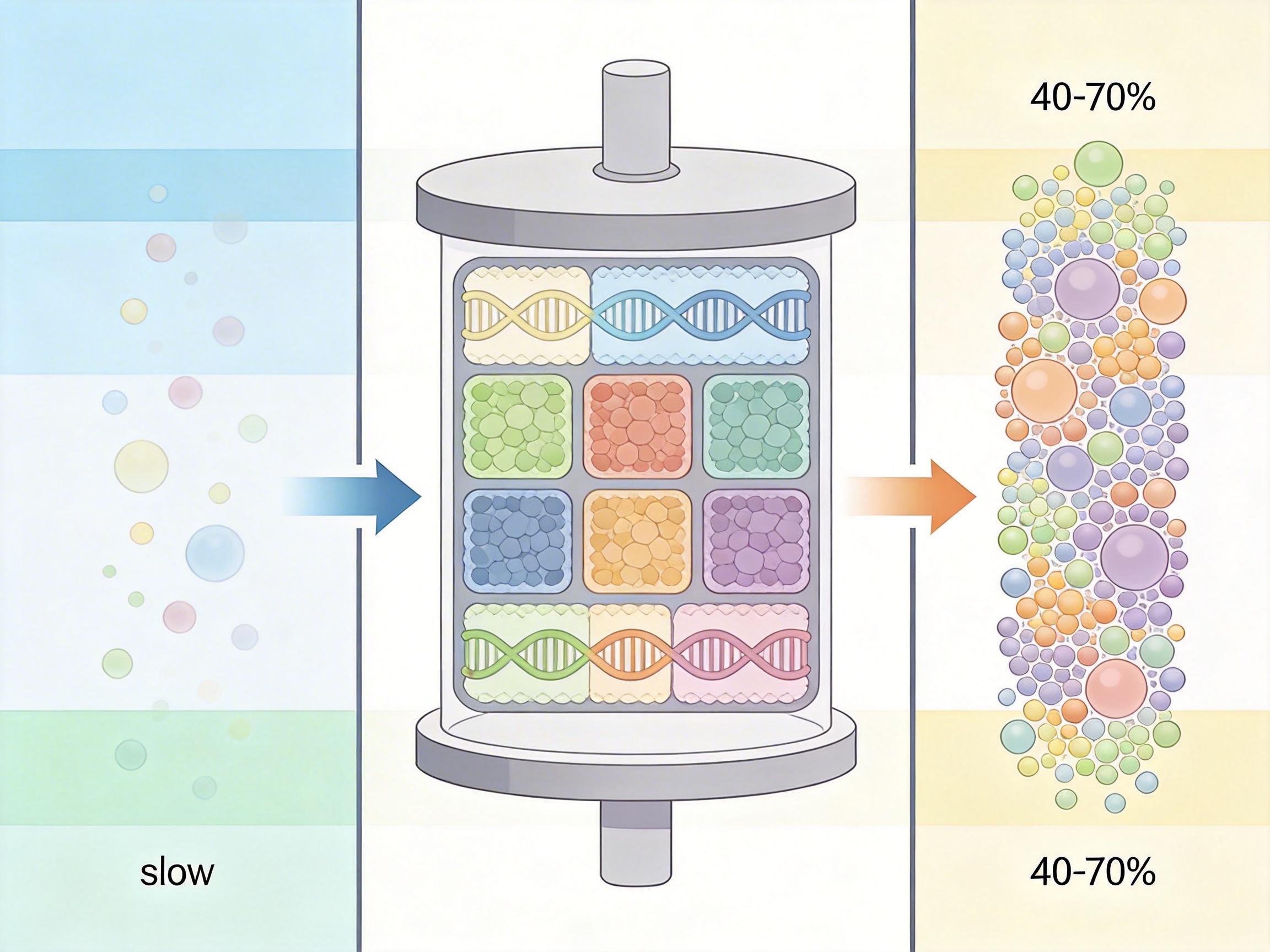
MULTI-evolve不是单点突破,而是一套从计算到实验的闭环系统,靠三个核心技术撑起来。
第一个支柱是**多模型集成的突变预测**。单一蛋白质语言模型(把氨基酸序列当“语言”学习规律的AI)总会有偏好,比如传统模型会排斥脯氨酸替换,就会错过APEX蛋白里能让活性提升53倍的A134P突变。MULTI-evolve把基于序列和结构的多个模型结合,还会修正模型对特定氨基酸的偏见,平均能找出20个有益突变,比单一模型多近一倍。
第二个支柱是神经网络的多突变外推。用单突变和双突变数据训练的全连接神经网络,能像资深厨师一样,从调料的搭配规律里,直接算出五种调料混合的味道。在12个数据集里,模型超过一半时间能精准选出功能最强的多突变体,实际实验里甚至只需要测9个候选就能找到最优解。
第三个支柱是**MULTI-assembly快速合成**。过去做多个突变的蛋白质合成,不仅贵还容易失败,MULTI-assembly通过优化引物设计和反应条件,对带9个突变的长序列能做到40%到70%的组装效率,把合成时间从数周压缩到数天。
很多人关注MULTI-evolve的效率提升,但我觉得更重要的是它代表的“实验室-循环”模式——计算和实验从一开始就绑定在一起,而不是先算完再去实验室验证。
传统的AI辅助生物研究,往往是计算团队先出预测,再扔给实验团队去试,两边像两条平行线。但Arc研究所的团队从一开始就让计算和实验人员一起工作:计算模型的训练目标完全围绕实验能测的200个样本设计,实验的每一组数据又直接喂给模型优化。这种模式下,AI不再是实验室的“外部顾问”,而是和实验仪器一样,成为研究的核心工具。
当然,这套方法也有局限:它目前更适合已经有部分功能基础的蛋白质,要从头设计全新功能的蛋白还得结合其他技术。但它至少解决了一个现实的痛点:当实验室只能测几百个样本时,怎么把每一份样本的价值用到极致。
MULTI-evolve的开源,让更多实验室能用上这套工具——未来可能会有更多酶、基因编辑工具、治疗性抗体,用这种方法快速优化升级。我们总说AI要赋能科学,但真正的赋能从来不是让AI代替人做决策,而是让人的每一份努力都能得到最精准的反馈。
从天文数字的序列空间里,靠200个样本找到最优解,这不是AI的魔法,而是人类终于学会了用更聪明的方式和自然对话。精准比海量更重要,协同比叠加更有效。