对抗知识焦虑,从看懂这条开始
App 下载
量子+超算联手,算出万原子级药物分子
电子相互作用|蛋白质分子模拟|日本理化学研究所|IBM|克利夫兰诊所|量子科学|新药研发|数理基础|医学健康
对抗知识焦虑,从看懂这条开始
App 下载电子相互作用|蛋白质分子模拟|日本理化学研究所|IBM|克利夫兰诊所|量子科学|新药研发|数理基础|医学健康
想象一下:在电脑里还原一个和真实细胞环境一模一样的蛋白质——12635个原子,每个都在水分子的包裹下振动、纠缠,连电子的细微波动都分毫不差。这不是科幻,是2026年5月,克利夫兰诊所、IBM和日本理化学研究所的团队刚完成的事。他们用两台量子计算机和两台超级计算机,把之前量子模拟的分子规模直接翻了40倍,精度提了210倍。而这一切,只是为了搞懂:一颗药,到底是怎么和我们体内的蛋白结合的?
要理解药物的疗效,得先搞懂药物分子和靶标蛋白的电子相互作用——这是经典计算机的死穴。
你可以把经典计算机的计算逻辑想象成查字典:每一个分子的电子状态都要对应一个预设的“词条”,但蛋白质里的原子动辄上万,电子之间的纠缠关系是指数级增长的,就像一本无限扩容的字典,经典计算机翻到报废也查不完。比如用传统的密度泛函理论模拟1万个原子的蛋白,需要的计算资源是现有全球超算总和的几十倍。
过去科学家只能用简化的“力场模型”——相当于给分子套上统一的“动作模板”,但这会漏掉关键的量子细节:比如某个电子的微小偏移,可能就决定了药物是有效还是有毒。
而量子计算机的逻辑是“直接模拟”——它本身就是用量子态来计算,能天然还原电子的纠缠和叠加。但现在的量子计算机还处在“噪声中间规模量子”(NISQ)阶段:量子比特少,还容易受干扰出错,单独用它连一个中等大小的分子都算不完。
这次突破的核心,是一套叫EWF-TrimSQD的混合算法——简单说就是“分工干活”。
你可以把整个蛋白质想象成一幅巨型拼图:经典超级计算机先把它拆成几百块小碎片,只留下那些电子纠缠最紧密的核心碎片——比如药物和蛋白结合的关键区域,交给量子计算机处理;剩下的“背景碎片”,比如周围的水分子,就用经典方法快速计算。
具体到操作:
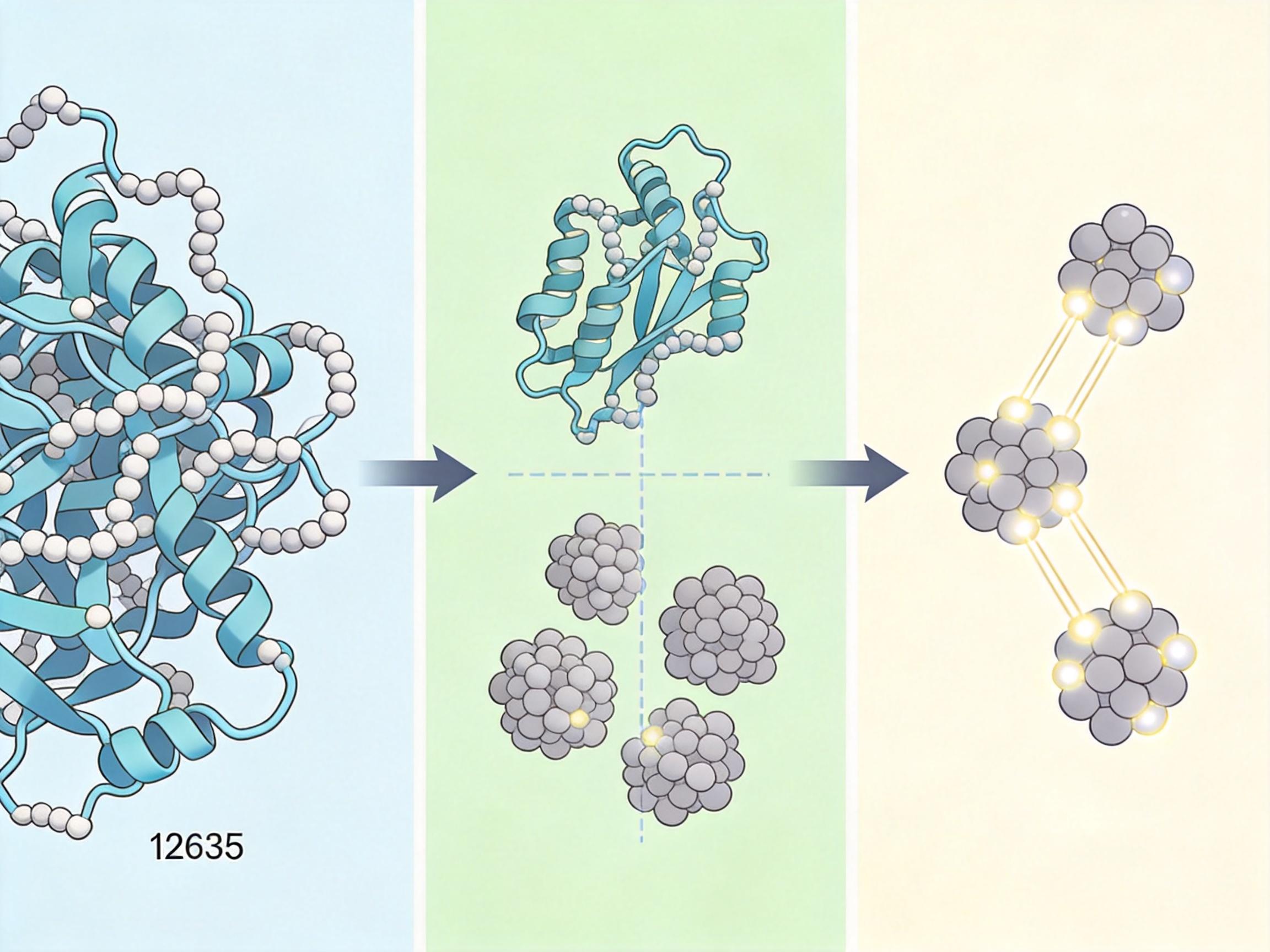
整个过程来回迭代了100多小时,量子计算机和超算像两个配合默契的工匠,各自干最擅长的活——这就是“量子中心超级计算”的核心逻辑:不用等完美的量子计算机,现在就能把现有工具的潜力榨到极致。

现在的成果,还只是“证明可行”,离真正的药物设计还有几道坎要跨。
首先是碎片划分的“艺术”:怎么拆蛋白才能既保证精度又不浪费计算资源?比如如果把药物结合的关键区域拆碎了,量子计算再精准也没用。目前团队用的是已经研究透的“标准蛋白”,换成未知结构的新型靶标,碎片划分的难度会呈指数级上升。
其次是量子误差的问题:现在的量子计算机还没有纠错能力,13亿次测量里有大量噪声数据,需要经典算法反复过滤。虽然这次的精度比半年前提升了210倍,但和经典计算的最高精度比,还有差距——目前只是“相当”,还没到“超越”。
最关键的是,量子优势还没被证明。也就是说,我们还不确定这种混合方法在所有蛋白体系里,都能比纯经典计算更快更准。匹兹堡大学的Junyu Liu就指出:“现在的成果很惊艳,但要证明量子计算在药物研发里的不可替代性,还需要更多数据。”
不过好消息是,现在的混合模式已经能解决实际问题了:比如模拟已知蛋白和药物的结合,验证经典计算的结果,或者在经典计算的“盲区”里找到新的结合位点。
当量子计算机的量子比特在低温舱里振动,当超级计算机的风扇转得能掀起风,它们正在干的事,其实是把“生命的语言”翻译成人类能懂的数字。
过去我们研发药物,像在黑匣子里摸鱼:靠试错,靠经验,靠运气。而量子-经典混合计算,相当于给黑匣子开了一扇小窗——虽然还看不清全貌,但已经能看到里面的鱼在怎么游。
量子不是要取代经典,而是要补上经典的盲区。 也许再过5年,我们就能用上量子计算设计的药物:它的分子结构经过1万次电子级别的模拟,副作用被提前排除,疗效被精准预测。而这一切的起点,就是2026年的今天,两台量子计算机和两台超算联手算出的那12635个原子。