对抗知识焦虑,从看懂这条开始
App 下载
FDA拟禁减肥神药散装配制,安全与低价的平衡破局
药品安全|503B外包设施|司美格鲁肽|GLP-1药物|FDA|新药研发|医学健康
对抗知识焦虑,从看懂这条开始
App 下载药品安全|503B外包设施|司美格鲁肽|GLP-1药物|FDA|新药研发|医学健康
2026年春,美国500万正在使用GLP-1类减肥与糖尿病药物的患者突然发现,他们依赖的低价替代渠道可能要消失了。4月底FDA抛出一则提案:将司美格鲁肽、替泽帕肽等热门药物的活性成分,从503B外包设施的散装配制清单中剔除。这意味着那些比品牌药便宜近一半的复合注射剂,很快将彻底退出市场——除非这些药物再次陷入全国性短缺。为什么在品牌药供应已经缓解的今天,FDA要突然切断这条低价路径?背后藏着一场持续了14年的药品安全与可及性的拉锯。
你可以把503B外包设施理解成「介于药房和药厂之间的特殊车间」——它不需要针对单个患者处方,就能批量生产无菌注射剂等药物,直接供给医院或药房。这个监管类别诞生于2013年,源头是一场惨烈的悲剧:2012年新英格兰复合药房(NECC)生产的污染类固醇注射剂,引发了全美真菌性脑膜炎爆发,最终导致64人死亡、751人感染。

正是这场灾难推动国会通过《药品质量与安全法案》,设立503B制度,要求这类批量配制设施必须遵守FDA的cGMP(现行良好生产规范),接受定期风险检查,还要报告不良事件。但它的核心特权是:当FDA批准的药品短缺时,503B可以直接用散装原料配制「复制药」,填补供应缺口。
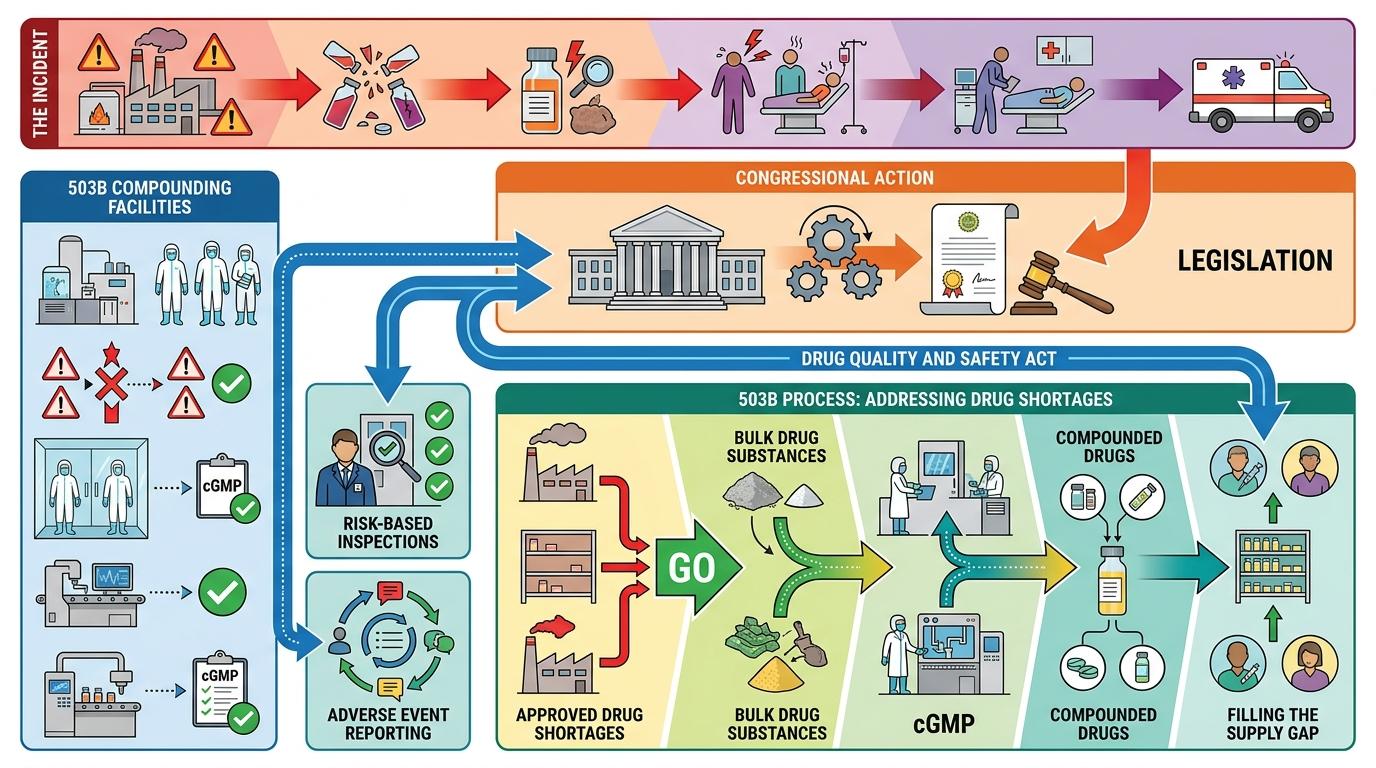
过去三年里,司美格鲁肽和替泽帕肽的全球需求暴涨了10倍,品牌药一度陷入持续短缺。这给了503B设施机会:它们用散装原料配制的复合注射剂,价格只有品牌药的40%-60%,迅速抢占了近20%的市场份额。但随着诺和诺德、礼来先后砸下数十亿美元扩产,2025年FDA宣布两类药物的短缺彻底解除——这场政策转向的导火索,就此点燃。
FDA在提案里说得很直接:「没有临床需求」让503B继续批量配制这些药物。这句话的潜台词是:既然品牌药能稳定供应,就没必要再让未经完整审批的复合药流通——毕竟复合药跳过了FDA对成品药的全套安全性验证,存在剂量不准、无菌不达标、成分纯度不足等风险。
数据能佐证这种担忧:截至2025年底,FDA已经收到超过1500例复合GLP-1药物的不良事件报告,其中17例直接死亡,不少案例都指向「活性成分含量不足导致疗效丧失」或「注射剂污染引发感染」。而品牌药的不良事件报告率,只有复合药的1/7。
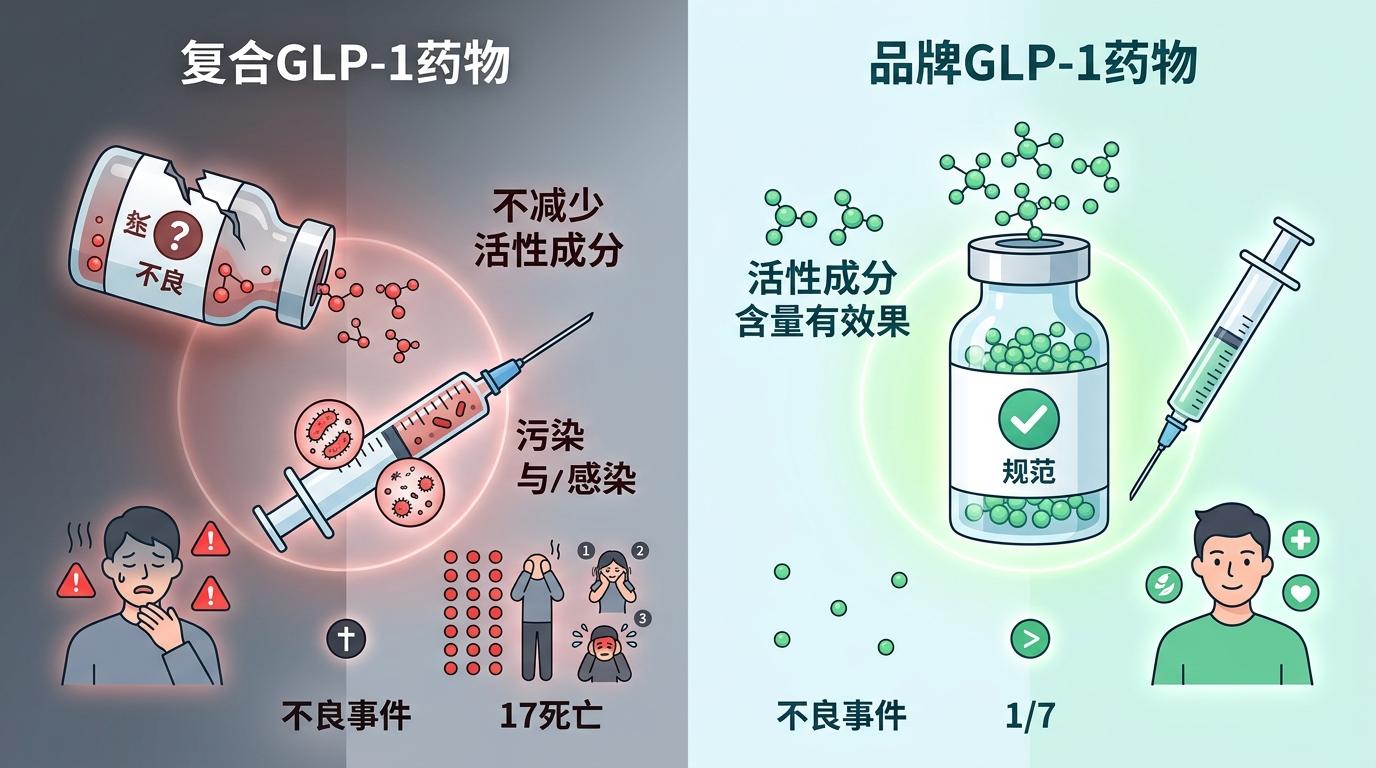
但反对声同样尖锐。美国有近30%的GLP-1使用者没有医保,品牌药每月1000-1300美元的费用,对他们来说是沉重负担。复合药的存在,相当于给了这些患者「用得起药」的选项。更现实的是,即便诺和诺德和礼来承诺降价,但品牌药的最终落地价格,仍可能比复合药高出30%以上。
FDA的选择,本质上是在「绝对安全」和「相对可及」之间踩刹车:它没有完全禁止复合药——针对单个患者处方的503A药房不受影响,只是堵死了「批量复制品牌药」的灰色地带。
对503B外包设施来说,这无疑是一记重拳。GLP-1类药物的配制业务,贡献了部分头部企业近40%的营收。失去这块业务后,它们要么转向罕见病、特殊剂型的定制配制,要么加大投入升级成更接近药厂的生产标准——但后者的合规成本,可能会让一半以上的中小型503B设施关门。
品牌药企则是直接受益者。诺和诺德和礼来过去两年扩产的产能,终于能完全释放到市场上。它们甚至已经提前出手:礼来宣布将Mounjaro的自付费用上限设为25美元/月,诺和诺德也推出了针对无保险患者的折扣计划——用可控的降价,彻底把复合药的生存空间挤掉。
最尴尬的是患者。短期内,那些依赖复合药的人要么被迫承担更高的药费,要么只能转向效果稍弱的旧款药物。长期来看,品牌药的竞争可能会推动价格进一步下降,但这个过程至少需要1-2年。而在这期间,部分患者可能会因为负担不起,中断治疗。
当FDA的提案在6月底结束公众意见征集时,这场关于安全与价格的争论还会持续更久。它暴露了一个更核心的问题:当一款药物从「救命刚需」变成「大众消费品」,监管该如何平衡商业利益与公共福祉?
安全永远是药品监管的底线,但可及性同样是不能被忽视的民生。「安全优先,但绝非唯一」,这或许是这场政策转向留给所有国家的启示。毕竟,再好的药物,如果普通人用不起,最终也只是橱窗里的展品。