对抗知识焦虑,从看懂这条开始
App 下载
FDA砍掉新药45%等待时间,靠实时数据监管
药物审批|临床试验流程|实时数据监管|Marty Makary|FDA|新药研发|医学健康
对抗知识焦虑,从看懂这条开始
App 下载药物审批|临床试验流程|实时数据监管|Marty Makary|FDA|新药研发|医学健康
当一个医生第1001次握着患者的手,说出“确诊癌症”这四个字时,他心里的疑问终于变成了行动。这位曾在约翰·霍普金斯医院主刀上千台手术的外科肿瘤医生,如今成了FDA专员——Marty Makary。他见过太多患者在等待新药的过程中耗尽生命,而传统新药研发里,从一期试验到最终提交审批,居然有45%的时间是“空转”的:没有试验在进行,只有繁琐的数据整理和文书往返。现在,他要把手术室里的实时监控逻辑搬到药物监管里——FDA将第一次像ICU医生盯患者 vitals 一样,盯着临床试验的每一个数据节点。为什么药物监管不能像救急诊一样快?这就是他给出的答案。
你可以把传统临床试验监管想象成:老师布置了作业,学生要等一个学期写完一本作业本,再统一交给老师批改——中间哪怕学生写不下去或者抄错了题,老师也完全不知道。而实时临床试验,就是老师坐在学生旁边,看着他每写一道题就检查一道。
这个转变的核心是一套基于云端和AI的数据流框架:试验数据从患者床旁、仪器终端实时同步到云端,AI会先做第一层筛选——自动识别异常的安全信号,比如患者突然发热、肝肾功能指标飙升,再把经过验证的核心数据同步给FDA的监管人员。阿斯利康的套细胞淋巴瘤试验已经用上了这套系统:当患者的肿瘤缩小数据刚出来,FDA的科学家就能在平台上看到,而不是等几个月后的试验报告。
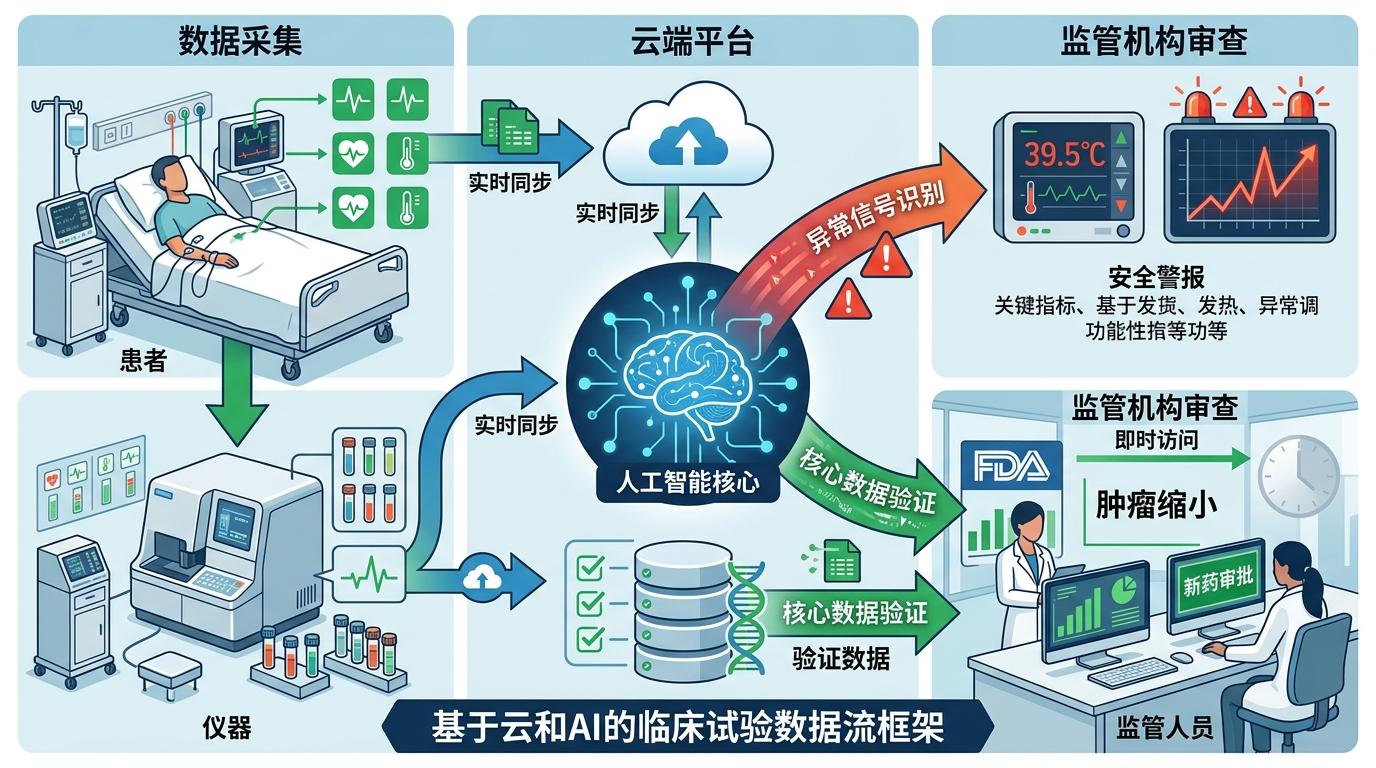
这套系统的关键不是“快”,而是“准”和“稳”。云端平台采用了合成数据和差分隐私技术——它会生成和真实数据统计特征完全一致的“假数据”供初步分析,既保护了患者隐私,又不耽误监管效率;AI算法则经过了超过3.8万项临床研究数据的训练,能精准区分“偶然的身体波动”和“需要警惕的不良反应”。
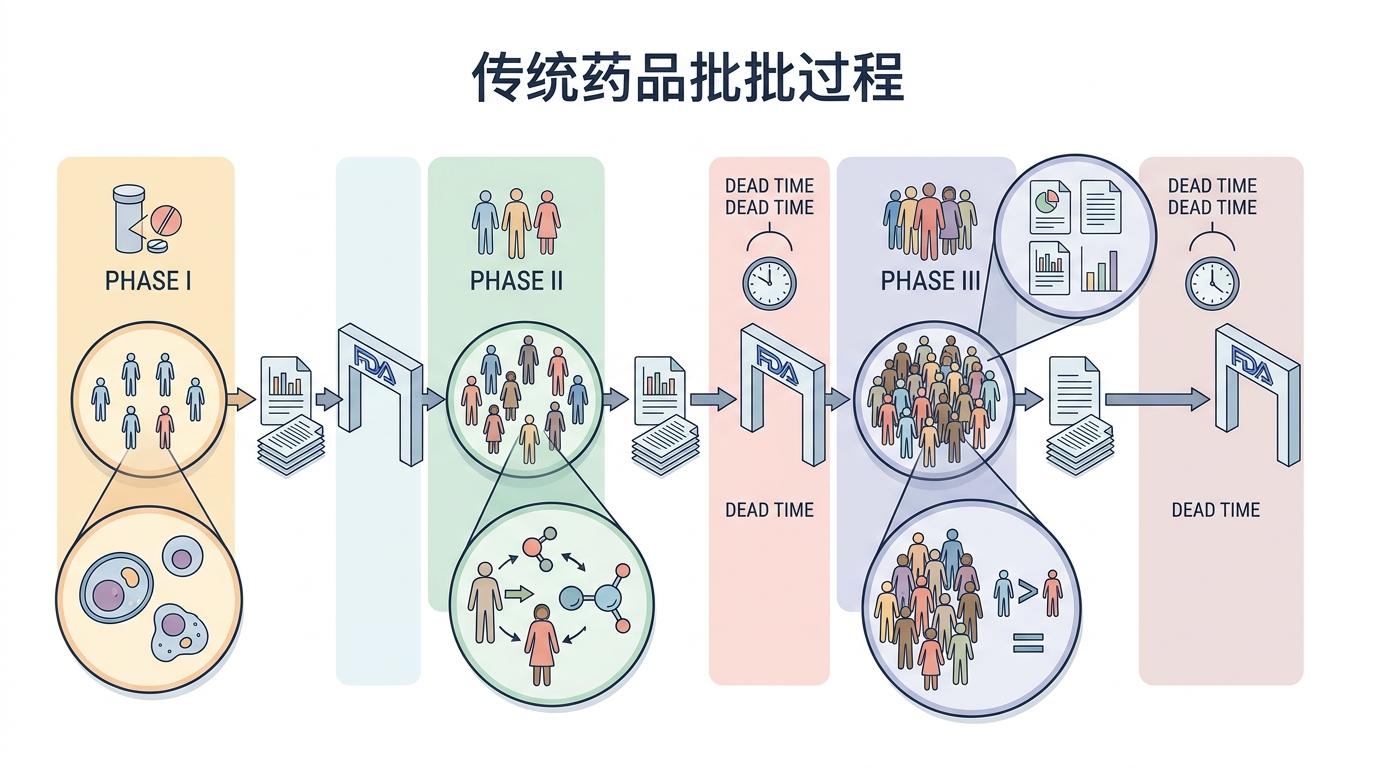
传统的分阶段审批逻辑,是把药物研发当成一个个独立的关卡:一期看安全,二期看疗效,三期看大规模验证,每过一关才能提交数据给FDA。这就像过收费站,每到一个站都要停车缴费、拿发票,大量时间耗在了“停车”上——也就是那些没有试验进行的“死时间”。
实时监管把这个“收费站模式”改成了“全程高速”。FDA不需要等一个阶段结束再看数据,而是在试验进行中就能持续监控:如果一款癌症药物在二期就显示出90%的肿瘤缩小率,监管人员可以提前介入,讨论是否提前启动三期或者加速审批;如果发现某个剂量组出现严重不良反应,也能立刻叫停,避免更多患者暴露在风险里。
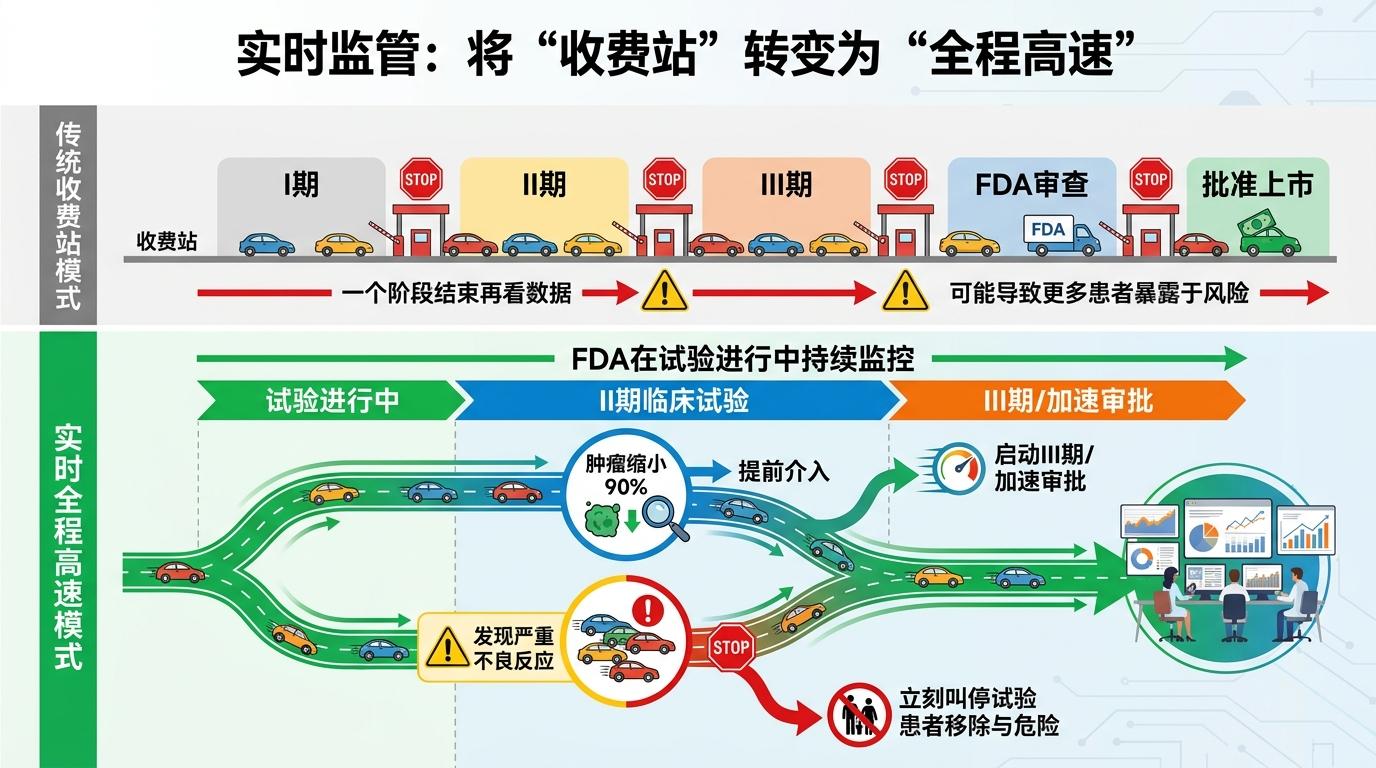
更值得关注的是,这不是监管方单方面的“提速”,而是整个临床试验生态的协同。FDA和药企共同制定实时数据的上报标准,比如哪些安全信号必须立刻上报,哪些疗效指标可以按周汇总;医院的研究人员也能通过平台实时看到数据反馈,及时调整试验操作。这种模式下,监管不再是研发的“终点审查”,而是全程的“动态护航”。
当然,风险也随之而来。加速审批会不会导致安全漏洞?FDA的回应是,实时监控反而能更早发现不良反应——就像ICU里的监护仪能在患者休克前就捕捉到血压下降的信号,比事后看报告要灵敏得多。目前的试点数据显示,实时监测到安全信号的时间,比传统模式平均提前了27天。
很多人把实时临床试验当成医药行业的“颠覆式创新”,但本质上,它只是把早已在ICU、民航、金融风控里普及的实时监控逻辑,搬到了药物研发领域。就像Marty Makary说的:“我在手术室里不会等几周后的报告才知道患者情况,为什么药物监管要等?”
这种“补位”的背后,是患者需求的倒逼。过去十年,FDA批准的新药里,有超过60%是通过加速审批通道上市的,其中不少是针对罕见病和晚期癌症的药物——患者等不起10年的研发周期。实时临床试验把这种“特例”变成了“常态”:未来,所有新药研发都可能采用这种“边做边看”的模式,而不是只有“突破性疗法”才能享受加速待遇。
但这也意味着新的挑战:监管人员要学会和AI一起工作,药企要重构数据管理流程,甚至连伦理审查都要适应“动态调整”的试验设计。比如,当试验因为实时数据而提前改变分组,怎么保证患者的知情同意是充分的?当AI筛选的安全信号出现误判,怎么平衡效率和严谨?这些问题没有现成答案,只能在试点中一步步摸索。
当我们谈论“新药研发提速”时,我们谈论的从来都不是冰冷的时间数字,而是一个个在等待中耗尽希望的患者。Marty Makary的那句疑问,最终变成了一套改变行业的规则:“为什么不能让患者更早拿到救命的药?”
实时临床试验不是要“放松监管”,而是要让监管更“聪明”——用技术把等待的时间压缩,把试错的成本降低,把救命的药更快递到患者手里。监管的终极目标,从来都是让生命等不起的人,不再等。
未来的某天,当一个患者拿到刚获批的新药时,他可能不会知道,这背后是一场从手术室到监管局的逻辑革命,但他会知道:自己不用再等那漫长的10年了。